

Infection and apoptosis as a combined inflammatory trigger
- PMID: 20137905
- PMCID: PMC5800876
- DOI: 10.1016/j.coi.2010.01.003
Infection and apoptosis as a combined inflammatory trigger
Abstract
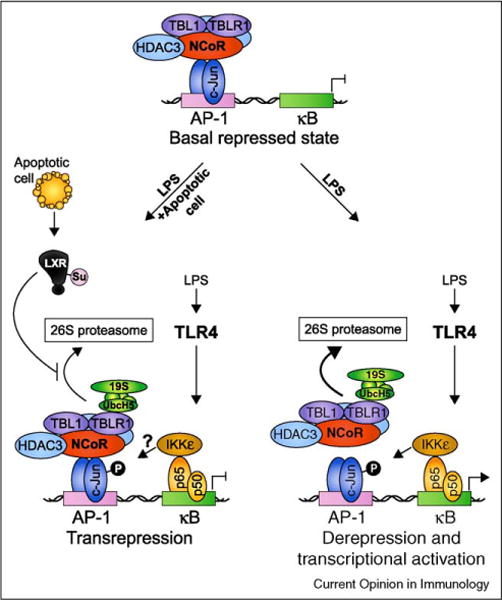
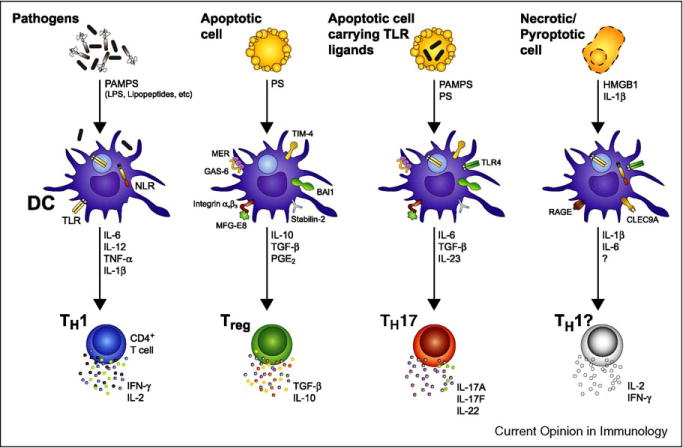
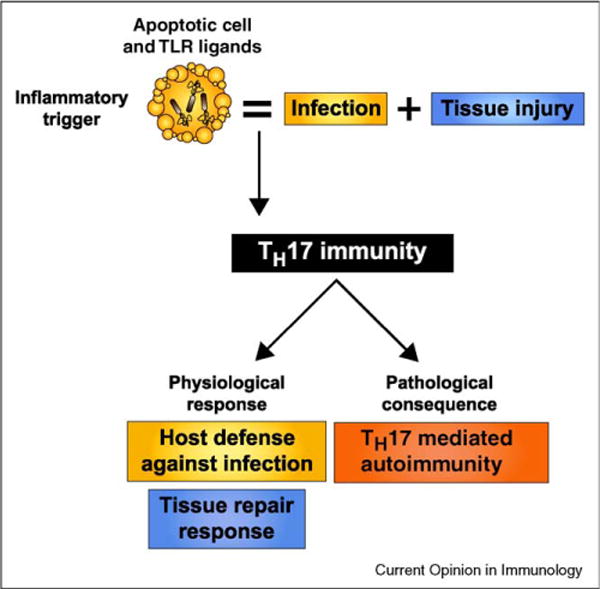
While inflammatory phagocytosis of microbial pathogens and non-inflammatory phagocytosis of apoptotic cells have each been studied extensively, the consequences of innate immune recognition of host cells undergoing apoptosis as a direct result of infection are unclear. In this situation, the innate immune system is confronted with mixed signals, those from apoptotic cells and those from the infecting pathogen. Nuclear receptor activation has been implicated downstream of apoptotic cell recognition while Toll-like receptors are the prototypical inflammatory receptors engaged during infection. When the two signals combine, a new set of events takes place beginning with transrepression of a subset of inflammatory-response genes and ending with the induction of a T helper-17 adaptive immune response. This response is best suited for clearing the infecting pathogen and repairing the damage that occurred to the host tissue during infection.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures
References
-
- Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96:245–254. - PubMed
-
- Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, et al. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. This paper showed that patients with SLE had apoptotic cell material in dendritic cells located in lymph node germinal centers, and autoantibodies that had gained autoreactivity in germinal center reactions. This supported the hypothesis that impaired clearance of apoptotic cells in patients with SLE could lead to secondary necrosis and buildup of self-antigens in tissues, contributing to autoimmunity in this disease. - PubMed
-
- Janko C, Schorn C, Grossmayer GE, Frey B, Herrmann M, Gaipl US, Munoz LE. Inflammatory clearance of apoptotic remnants in systemic lupus erythematosus (SLE) Autoimmun Rev. 2008;8:9–12. - PubMed
-
- Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
