

RecQ-dependent death-by-recombination in cells lacking RecG and UvrD
- PMID: 20138014
- PMCID: PMC2846991
- DOI: 10.1016/j.dnarep.2009.12.019
RecQ-dependent death-by-recombination in cells lacking RecG and UvrD
Abstract
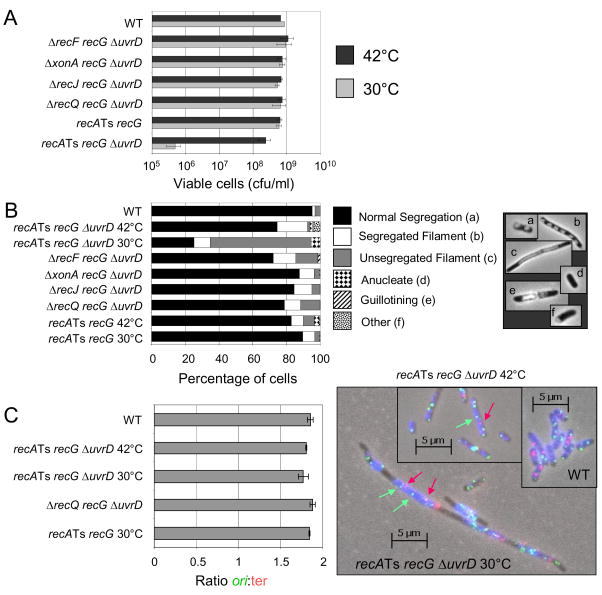
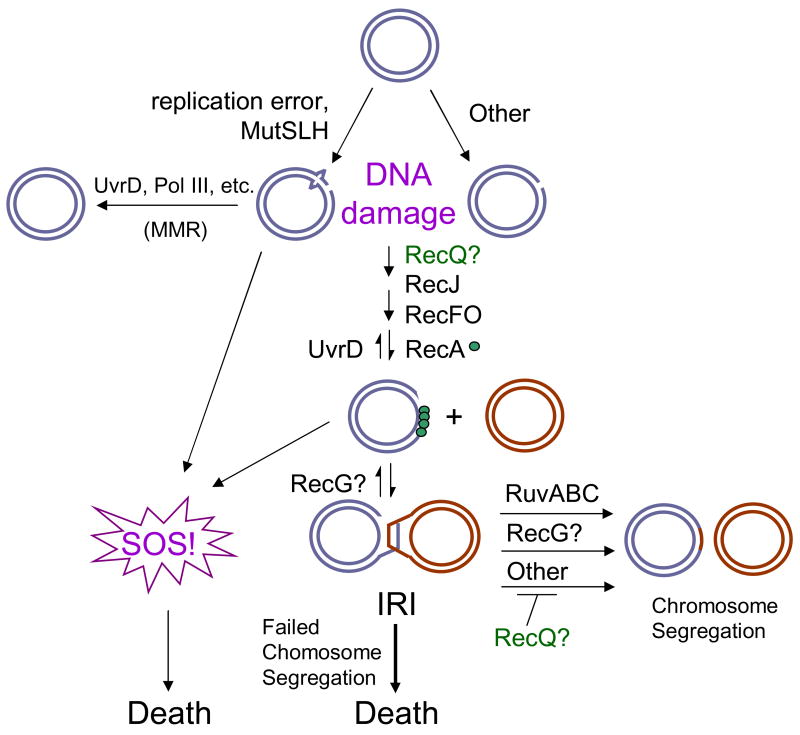
Maintenance of genomic stability is critical for all cells. Homologous recombination (HR) pathways promote genome stability using evolutionarily conserved proteins such as RecA, SSB, and RecQ, the Escherichia coli homologue of five human proteins at least three of which suppress genome instability and cancer. A previous report indicated that RecQ promotes the net accumulation in cells of intermolecular HR intermediates (IRIs), a net effect opposite that of the yeast and two human RecQ homologues. Here we extend those conclusions. We demonstrate that cells that lack both UvrD, an inhibitor of RecA-mediated strand exchange, and RecG, a DNA helicase implicated in IRI resolution, are inviable. We show that the uvrD recG cells die a "death-by-recombination" in which IRIs accumulate blocking chromosome segregation. First, their death requires RecA HR protein. Second, the death is accompanied by cytogenetically visible failure to segregate chromosomes. Third, FISH analyses show that the unsegregated chromosomes have completed replication, supporting the hypothesis that unresolved IRIs prevented the segregation. Fourth, we show that RecQ and induction of the SOS response are required for the accumulation of replicated, unsegregated chromosomes and death, as are RecF, RecO, and RecJ. ExoI exonuclease and MutL mismatch-repair protein are partially required. This set of genes is similar but not identical to those that promote death-by-recombination of DeltauvrD Deltaruv cells. The data support models in which RecQ promotes the net accumulation in cells of IRIs and RecG promotes resolution of IRIs that form via pathways not wholly identical to those that produce the IRIs resolved by RuvABC. This implies that RecG resolves intermediates other than or in addition to standard Holliday junctions resolved by RuvABC. The role of RecQ in net accumulation of IRIs may be shared by one or more of its human homologues.
2009 Elsevier B.V. All rights reserved.
Figures
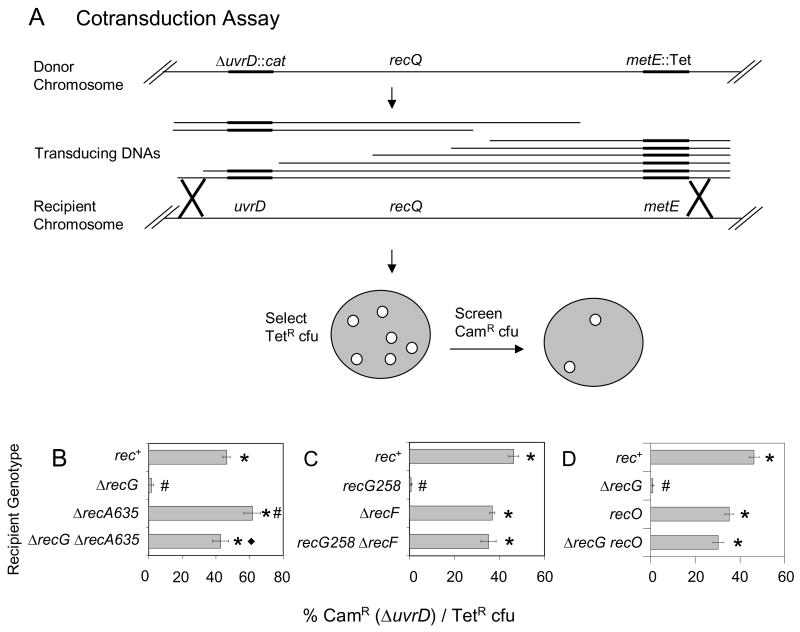
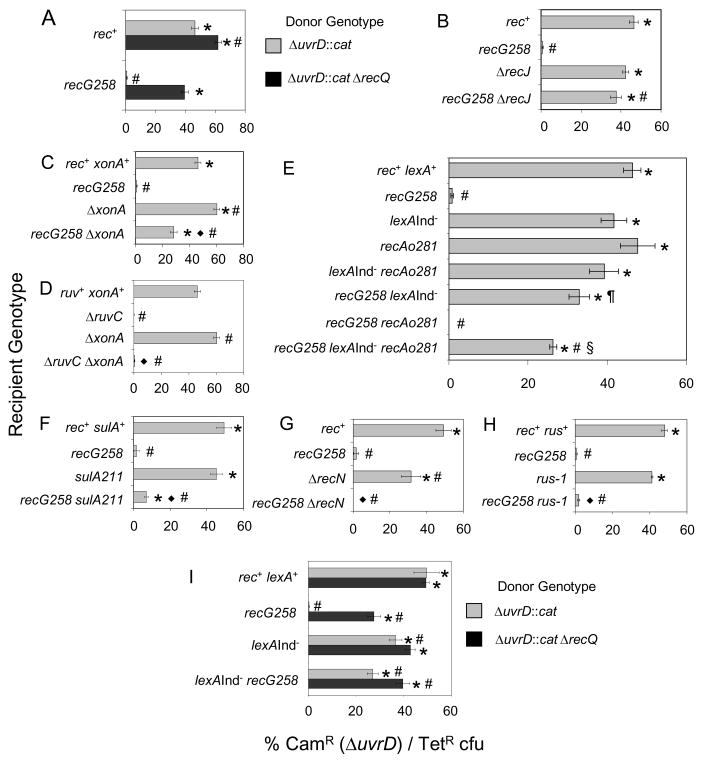
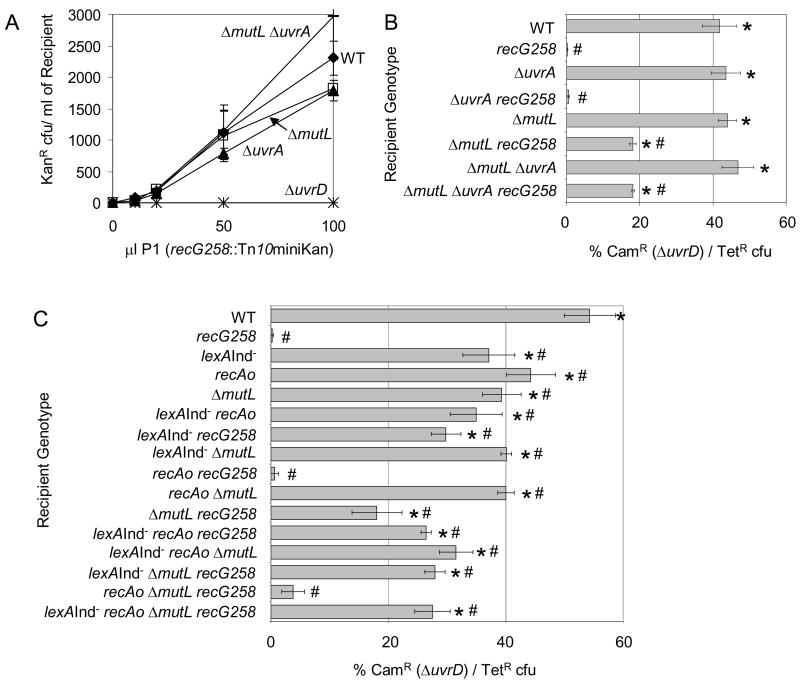
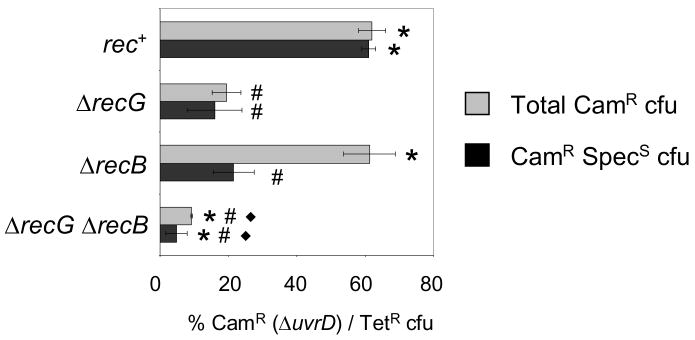
Similar articles
-
RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins.Mol Cell. 2007 Apr 27;26(2):273-86. doi: 10.1016/j.molcel.2007.03.012. Mol Cell. 2007. PMID: 17466628 Free PMC article.
-
Pathways of resistance to thymineless death in Escherichia coli and the function of UvrD.Genetics. 2011 Sep;189(1):23-36. doi: 10.1534/genetics.111.130161. Epub 2011 Jul 29. Genetics. 2011. PMID: 21705756 Free PMC article.
-
RecQ helicase acts before RuvABC, RecG and XerC proteins during recombination in recBCD sbcBC mutants of Escherichia coli.Res Microbiol. 2013 Dec;164(10):987-97. doi: 10.1016/j.resmic.2013.08.008. Epub 2013 Sep 12. Res Microbiol. 2013. PMID: 24036154
-
Functions of RecQ family helicases: possible involvement of Bloom's and Werner's syndrome gene products in guarding genome integrity during DNA replication.J Biochem. 2001 Apr;129(4):501-7. doi: 10.1093/oxfordjournals.jbchem.a002883. J Biochem. 2001. PMID: 11275547 Review.
-
Biochemistry of homologous recombination in Escherichia coli.Microbiol Rev. 1994 Sep;58(3):401-65. doi: 10.1128/mr.58.3.401-465.1994. Microbiol Rev. 1994. PMID: 7968921 Free PMC article. Review.
Cited by
-
PcrA-mediated disruption of RecA nucleoprotein filaments--essential role of the ATPase activity of RecA.Nucleic Acids Res. 2012 Sep 1;40(17):8416-24. doi: 10.1093/nar/gks641. Epub 2012 Jun 28. Nucleic Acids Res. 2012. PMID: 22743269 Free PMC article.
-
Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase.Sci Adv. 2016 Nov 18;2(11):e1601605. doi: 10.1126/sciadv.1601605. eCollection 2016 Nov. Sci Adv. 2016. PMID: 28090586 Free PMC article.
-
Identification of recG genetic interactions in Escherichia coli by transposon sequencing.J Bacteriol. 2023 Dec 19;205(12):e0018423. doi: 10.1128/jb.00184-23. Epub 2023 Nov 29. J Bacteriol. 2023. PMID: 38019006 Free PMC article.
-
Interaction with the carboxy-terminal tip of SSB is critical for RecG function in E. coli.Nucleic Acids Res. 2023 May 8;51(8):3735-3753. doi: 10.1093/nar/gkad162. Nucleic Acids Res. 2023. PMID: 36912097 Free PMC article.
-
Genetic analysis of Escherichia coli RadA: functional motifs and genetic interactions.Mol Microbiol. 2015 Mar;95(5):769-79. doi: 10.1111/mmi.12899. Epub 2015 Jan 30. Mol Microbiol. 2015. PMID: 25484163 Free PMC article.
References
-
- Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, Jasin M, Vogel H, Sung P, Luo G. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007;21:3073–3084. - PMC - PubMed
-
- Friedberg E, Walker G, Siede W, Wood R, Schultz R, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D.C: 2005.
-
- Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34:264–272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
