

Non-muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury
- PMID: 20139351
- PMCID: PMC3028257
- DOI: 10.1165/rcmb.2009-0197OC
Non-muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury
Abstract
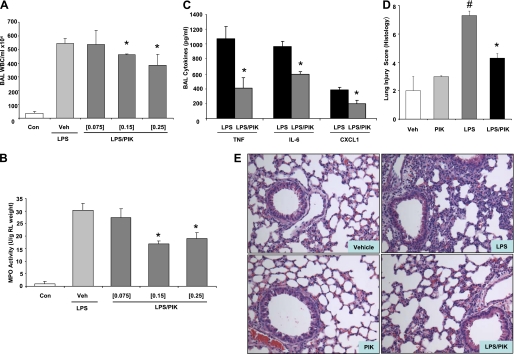
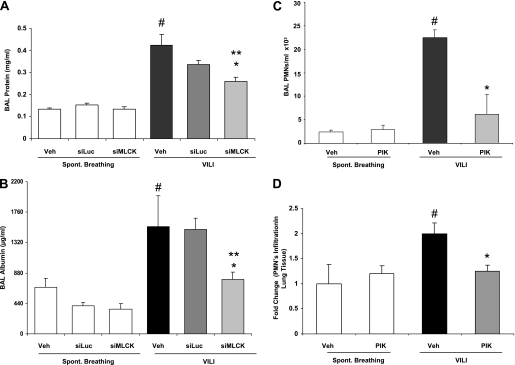
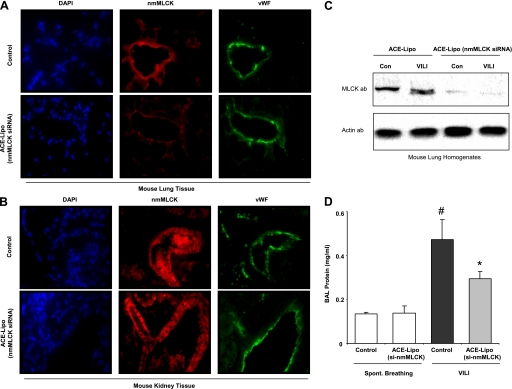
Acute lung injury (ALI) and mechanical ventilator-induced lung injury (VILI), major causes of acute respiratory failure with elevated morbidity and mortality, are characterized by significant pulmonary inflammation and alveolar/vascular barrier dysfunction. Previous studies highlighted the role of the non-muscle myosin light chain kinase isoform (nmMLCK) as an essential element of the inflammatory response, with variants in the MYLK gene that contribute to ALI susceptibility. To define nmMLCK involvement further in acute inflammatory syndromes, we used two murine models of inflammatory lung injury, induced by either an intratracheal administration of lipopolysaccharide (LPS model) or mechanical ventilation with increased tidal volumes (the VILI model). Intravenous delivery of the membrane-permeant MLC kinase peptide inhibitor, PIK, produced a dose-dependent attenuation of both LPS-induced lung inflammation and VILI (~50% reductions in alveolar/vascular permeability and leukocyte influx). Intravenous injections of nmMLCK silencing RNA, either directly or as cargo within angiotensin-converting enzyme (ACE) antibody-conjugated liposomes (to target the pulmonary vasculature selectively), decreased nmMLCK lung expression (∼70% reduction) and significantly attenuated LPS-induced and VILI-induced lung inflammation (∼40% reduction in bronchoalveolar lavage protein). Compared with wild-type mice, nmMLCK knockout mice were significantly protected from VILI, with significant reductions in VILI-induced gene expression in biological pathways such as nrf2-mediated oxidative stress, coagulation, p53-signaling, leukocyte extravasation, and IL-6-signaling. These studies validate nmMLCK as an attractive target for ameliorating the adverse effects of dysregulated lung inflammation.
Figures





References
-
- Lazar V, Garcia JG. A single human myosin light chain kinase gene (MLCK; MYLK) transcribes multiple nonmuscle isoforms. Genomics 1999;57:256–267. - PubMed
-
- Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998;157:294–323. - PubMed
-
- Ma SF, Grigoryev DN, Taylor AD, Nonas S, Sammani S, Ye SQ, Garcia JG. Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol 2005;289:L468–L477. - PubMed
-
- Chopra M, Reuben JS, Sharma AC. Acute lung injury: apoptosis and signaling mechanisms. Exp Biol Med 2009;234:361–371. - PubMed
-
- Petrache I, Verin AD, Crow MT, Birukova A, Liu F, Garcia JG. Differential effect of MLC kinase in TNF-alpha–induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol 2001;280:L1168–L1178. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
