

Advanced identification of proteins in uncharacterized proteomes by pulsed in vivo stable isotope labeling-based mass spectrometry
- PMID: 20139370
- PMCID: PMC2877977
- DOI: 10.1074/mcp.M900426-MCP200
Advanced identification of proteins in uncharacterized proteomes by pulsed in vivo stable isotope labeling-based mass spectrometry
Abstract
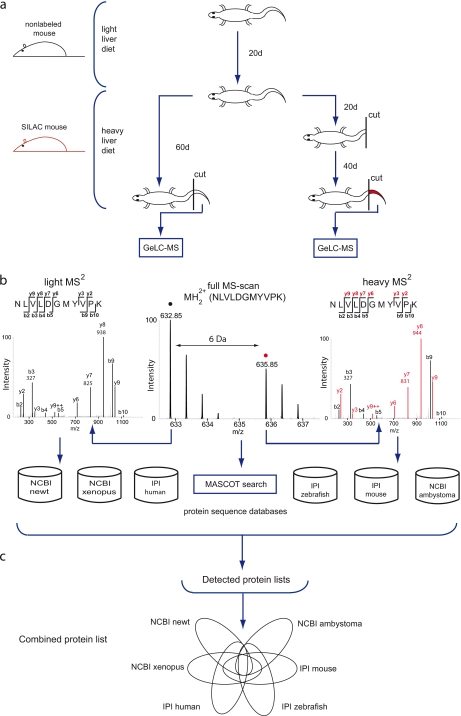
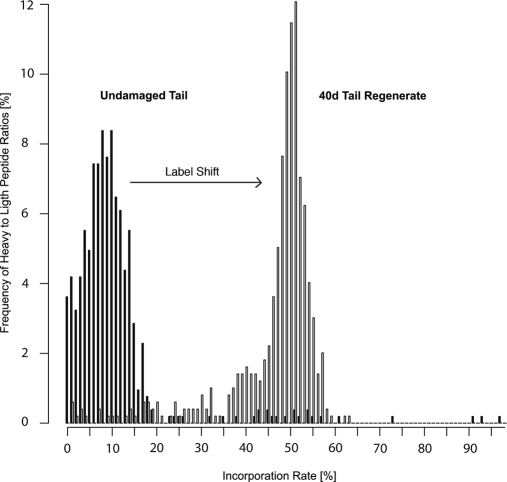
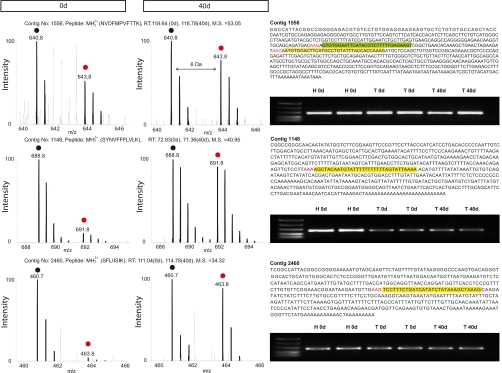
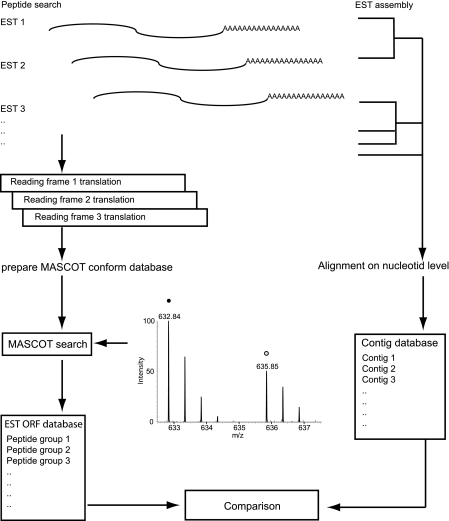
Despite progress in the characterization of their genomes, proteomes of several model organisms are often only poorly characterized. This problem is aggravated by the presence of large numbers of expressed sequence tag clones that lack homologues in other species, which makes it difficult to identify new proteins irrespective of whether such molecules are involved in species-specific biological processes. We have used a pulsed stable isotope labeling with amino acids in cell culture (SILAC)-based mass spectrometry method, which is based on the detection of paired peptides after [(13)C(6)]lysine incorporation into proteins in vivo, to greatly increase the confidence of protein identification in cross-species database searches. The method was applied to identify nearly 3000 proteins in regenerating tails of the urodele amphibian Notophthalmus viridescens, which possesses outstanding capabilities in the regeneration of complex tissues. We reason that pulsed in vivo SILAC represents a versatile tool to identify new proteins in species for which only limited sequence information exists.
Figures

Similar articles
-
Spiked-in pulsed in vivo labeling identifies a new member of the CCN family in regenerating newt hearts.J Proteome Res. 2012 Sep 7;11(9):4693-704. doi: 10.1021/pr300521p. Epub 2012 Aug 27. J Proteome Res. 2012. PMID: 22891955
-
Stable isotope labeling of Arabidopsis thaliana cells and quantitative proteomics by mass spectrometry.Mol Cell Proteomics. 2005 Nov;4(11):1697-709. doi: 10.1074/mcp.M500190-MCP200. Epub 2005 Aug 8. Mol Cell Proteomics. 2005. PMID: 16088002
-
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) Technology in Fission Yeast.Cold Spring Harb Protoc. 2017 Jun 1;2017(6):pdb.top079814. doi: 10.1101/pdb.top079814. Cold Spring Harb Protoc. 2017. PMID: 28572211
-
Proteome analysis by mass spectrometry.Annu Rev Biophys Biomol Struct. 2003;32:399-424. doi: 10.1146/annurev.biophys.32.110601.141854. Epub 2003 Jan 28. Annu Rev Biophys Biomol Struct. 2003. PMID: 12574065 Review.
-
Advances in quantitative proteomics via stable isotope tagging and mass spectrometry.Curr Opin Biotechnol. 2003 Feb;14(1):110-8. doi: 10.1016/s0958-1669(02)00018-6. Curr Opin Biotechnol. 2003. PMID: 12566010 Review.
Cited by
-
The SILAC fly allows for accurate protein quantification in vivo.Mol Cell Proteomics. 2010 Oct;9(10):2173-83. doi: 10.1074/mcp.M110.000323. Epub 2010 Jun 5. Mol Cell Proteomics. 2010. PMID: 20525996 Free PMC article.
-
Advancements in Oncoproteomics Technologies: Treading toward Translation into Clinical Practice.Proteomes. 2023 Jan 10;11(1):2. doi: 10.3390/proteomes11010002. Proteomes. 2023. PMID: 36648960 Free PMC article. Review.
-
Molecular signatures that correlate with induction of lens regeneration in newts: lessons from proteomic analysis.Hum Genomics. 2014 Dec 11;8(1):22. doi: 10.1186/s40246-014-0022-y. Hum Genomics. 2014. PMID: 25496664 Free PMC article.
-
A de novo assembly of the newt transcriptome combined with proteomic validation identifies new protein families expressed during tissue regeneration.Genome Biol. 2013 Feb 20;14(2):R16. doi: 10.1186/gb-2013-14-2-r16. Genome Biol. 2013. PMID: 23425577 Free PMC article.
-
Exploring ribosome composition and newly synthesized proteins through proteomics and potential biomedical applications.Expert Rev Proteomics. 2017 Jun;14(6):529-543. doi: 10.1080/14789450.2017.1333424. Epub 2017 May 26. Expert Rev Proteomics. 2017. PMID: 28532181 Free PMC article. Review.
References
-
- Yates J. R., 3rd (1998) Database searching using mass spectrometry data. Electrophoresis 19, 893–900 - PubMed
-
- Choudhary J. S., Blackstock W. P., Creasy D. M., Cottrell J. S. (2001) Matching peptide mass spectra to EST and genomic DNA databases. Trends Biotechnol 19, S17–S22 - PubMed
-
- Habermann B., Oegema J., Sunyaev S., Shevchenko A. (2004) The power and the limitations of cross-species protein identification by mass spectrometry-driven sequence similarity searches. Mol. Cell. Proteomics 3, 238–249 - PubMed
-
- Standing K. G. (2003) Peptide and protein de novo sequencing by mass spectrometry. Curr. Opin. Struct. Biol 13, 595–601 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
