

Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato
- PMID: 20140239
- PMCID: PMC2816678
- DOI: 10.1371/journal.pgen.1000796
Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato
Abstract
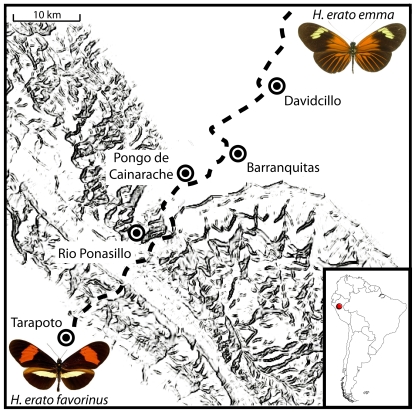
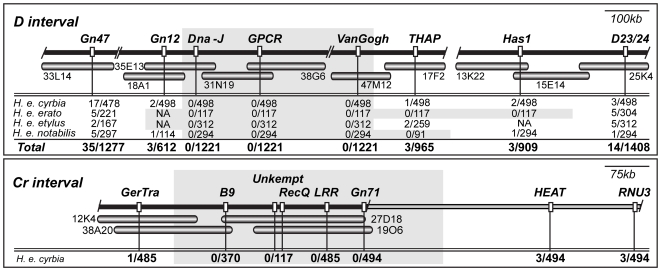
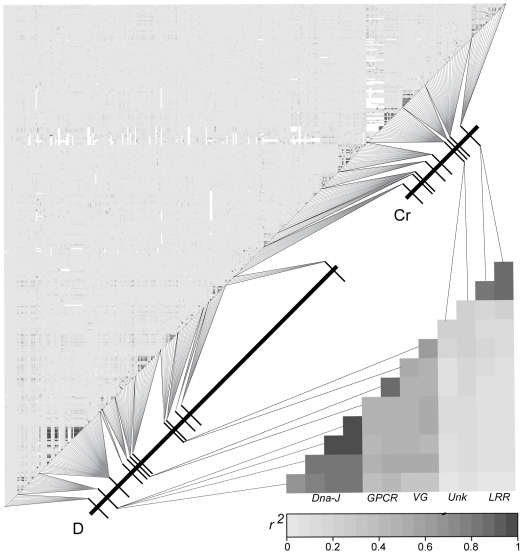
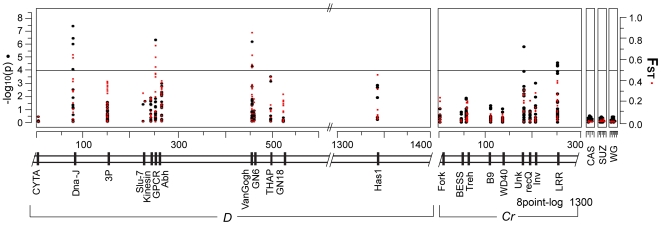
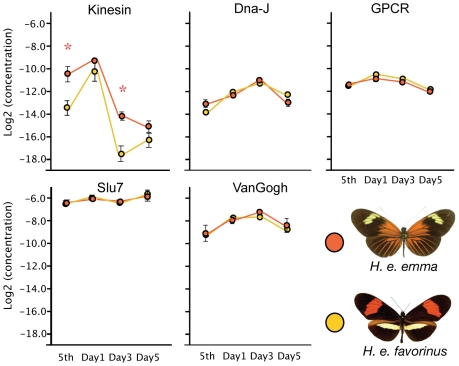
Wing pattern evolution in Heliconius butterflies provides some of the most striking examples of adaptation by natural selection. The genes controlling pattern variation are classic examples of Mendelian loci of large effect, where allelic variation causes large and discrete phenotypic changes and is responsible for both convergent and highly divergent wing pattern evolution across the genus. We characterize nucleotide variation, genotype-by-phenotype associations, linkage disequilibrium (LD), and candidate gene expression patterns across two unlinked genomic intervals that control yellow and red wing pattern variation among mimetic forms of Heliconius erato. Despite very strong natural selection on color pattern, we see neither a strong reduction in genetic diversity nor evidence for extended LD across either patterning interval. This observation highlights the extent that recombination can erase the signature of selection in natural populations and is consistent with the hypothesis that either the adaptive radiation or the alleles controlling it are quite old. However, across both patterning intervals we identified SNPs clustered in several coding regions that were strongly associated with color pattern phenotype. Interestingly, coding regions with associated SNPs were widely separated, suggesting that color pattern alleles may be composed of multiple functional sites, conforming to previous descriptions of these loci as "supergenes." Examination of gene expression levels of genes flanking these regions in both H. erato and its co-mimic, H. melpomene, implicate a gene with high sequence similarity to a kinesin as playing a key role in modulating pattern and provides convincing evidence for parallel changes in gene regulation across co-mimetic lineages. The complex genetic architecture at these color pattern loci stands in marked contrast to the single casual mutations often identified in genetic studies of adaptation, but may be more indicative of the type of genetic changes responsible for much of the adaptive variation found in natural populations.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Barrett RDH, Schluter D. Adaptation from standing genetic variation. Trends in Ecology & Evolution. 2008;23:38–44. - PubMed
-
- Colosimo PF, Hosemann KE, Balabhadra S, Villareal G, Dickson M, et al. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodyplasin alleles. Science. 2005;307:1928–1933. - PubMed
-
- Werner JD, Borevitz JO, Warthmann N, Trainer GT, Ecker JR, et al. Quantitative trait locus mapping and DNA array hybridization identify an FLM deletion as a cause for natural flowering-time variation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2460–2465. - PMC - PubMed
-
- Steiner CC, Weber JN, Hoekstra HE. Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biol. 2007;5:e219. doi: 10.1371/journal.pbio.0050219. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
