

Complete-proteome mapping of human influenza A adaptive mutations: implications for human transmissibility of zoonotic strains
- PMID: 20140252
- PMCID: PMC2815782
- DOI: 10.1371/journal.pone.0009025
Complete-proteome mapping of human influenza A adaptive mutations: implications for human transmissibility of zoonotic strains
Abstract
Background: There is widespread concern that H5N1 avian influenza A viruses will emerge as a pandemic threat, if they become capable of human-to-human (H2H) transmission. Avian strains lack this capability, which suggests that it requires important adaptive mutations. We performed a large-scale comparative analysis of proteins from avian and human strains, to produce a catalogue of mutations associated with H2H transmissibility, and to detect their presence in avian isolates.
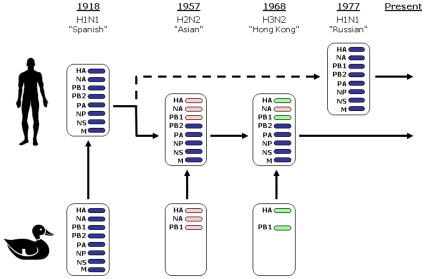
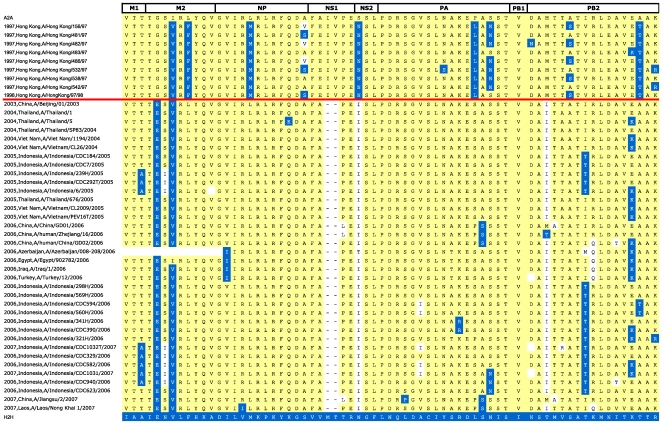
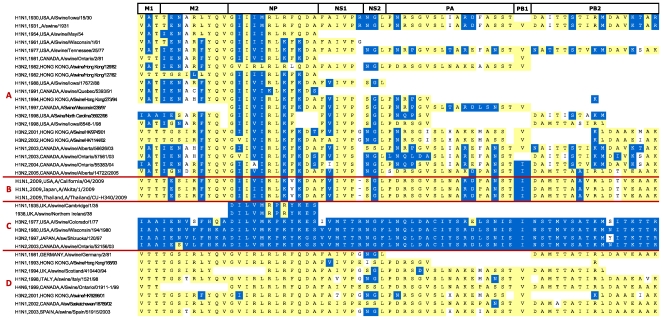
Methodology/principal findings: We constructed a dataset of influenza A protein sequences from 92,343 public database records. Human and avian sequence subsets were compared, using a method based on mutual information, to identify characteristic sites where human isolates present conserved mutations. The resulting catalogue comprises 68 characteristic sites in eight internal proteins. Subtype variability prevented the identification of adaptive mutations in the hemagglutinin and neuraminidase proteins. The high number of sites in the ribonucleoprotein complex suggests interdependence between mutations in multiple proteins. Characteristic sites are often clustered within known functional regions, suggesting their functional roles in cellular processes. By isolating and concatenating characteristic site residues, we defined adaptation signatures, which summarize the adaptive potential of specific isolates. Most adaptive mutations emerged within three decades after the 1918 pandemic, and have remained remarkably stable thereafter. Two lineages with stable internal protein constellations have circulated among humans without reassorting. On the contrary, H5N1 avian and swine viruses reassort frequently, causing both gains and losses of adaptive mutations.
Conclusions: Human host adaptation appears to be complex and systemic, involving nearly all influenza proteins. Adaptation signatures suggest that the ability of H5N1 strains to infect humans is related to the presence of an unusually high number of adaptive mutations. However, these mutations appear unstable, suggesting low pandemic potential of H5N1 in its current form. In addition, adaptation signatures indicate that pandemic H1N1/09 strain possesses multiple human-transmissibility mutations, though not an unusually high number with respect to swine strains that infected humans in the past. Adaptation signatures provide a novel tool for identifying zoonotic strains with the potential to infect humans.
Conflict of interest statement
Figures


Comment in
-
Proteomics search of influenza A viruses for adaptive mutations to human hosts.Expert Rev Proteomics. 2010 Jun;7(3):323-6. doi: 10.1586/epr.10.28. Expert Rev Proteomics. 2010. PMID: 20536303 No abstract available.
References
-
- Potter CW. A history of influenza. J Appl Microbiol. 2001;91:572–579. - PubMed
-
- Capua I, Alexander DJ. Avian influenza and human health. Acta Trop. 2002;83(1):1–6. - PubMed
-
- Peiris JS, Tu WW, Yen HL. A novel H1N1 virus causes the first pandemic of the 21(st) Century. Eur J Immunol (Epub ahead of print, Sep 29) 2009 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
