

Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics
- PMID: 20142485
- PMCID: PMC2840111
- DOI: 10.1073/pnas.0911295107
Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics
Abstract
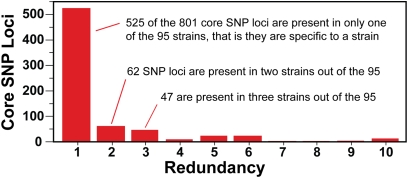
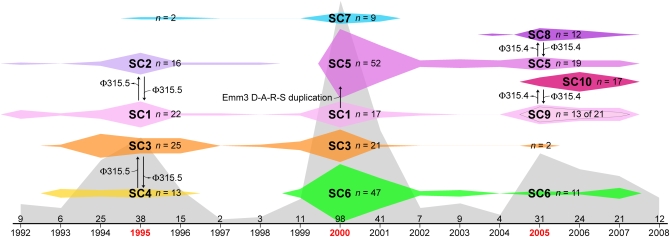
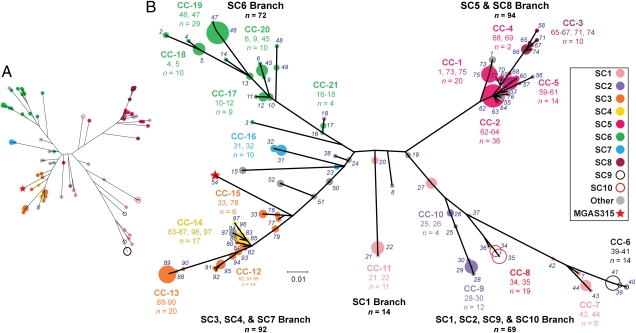
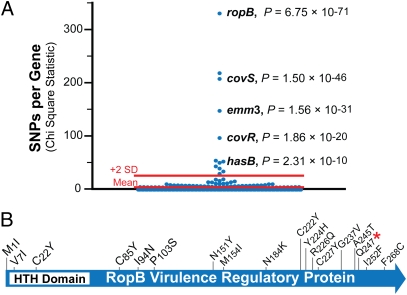
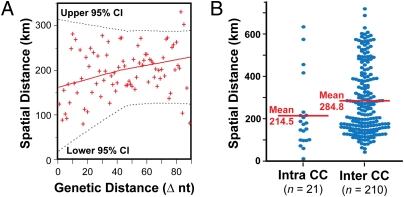
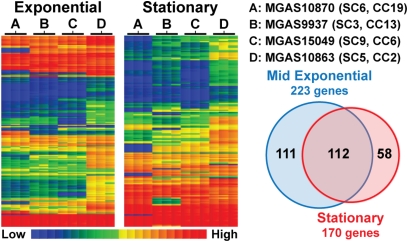
Understanding the fine-structure molecular architecture of bacterial epidemics has been a long-sought goal of infectious disease research. We used short-read-length DNA sequencing coupled with mass spectroscopy analysis of SNPs to study the molecular pathogenomics of three successive epidemics of invasive infections involving 344 serotype M3 group A Streptococcus in Ontario, Canada. Sequencing the genome of 95 strains from the three epidemics, coupled with analysis of 280 biallelic SNPs in all 344 strains, revealed an unexpectedly complex population structure composed of a dynamic mixture of distinct clonally related complexes. We discovered that each epidemic is dominated by micro- and macrobursts of multiple emergent clones, some with distinct strain genotype-patient phenotype relationships. On average, strains were differentiated from one another by only 49 SNPs and 11 insertion-deletion events (indels) in the core genome. Ten percent of SNPs are strain specific; that is, each strain has a unique genome sequence. We identified nonrandom temporal-spatial patterns of strain distribution within and between the epidemic peaks. The extensive full-genome data permitted us to identify genes with significantly increased rates of nonsynonymous (amino acid-altering) nucleotide polymorphisms, thereby providing clues about selective forces operative in the host. Comparative expression microarray analysis revealed that closely related strains differentiated by seemingly modest genetic changes can have significantly divergent transcriptomes. We conclude that enhanced understanding of bacterial epidemics requires a deep-sequencing, geographically centric, comparative pathogenomics strategy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- MacLean D, Jones JD, Studholme DJ. Application of ‘next-generation’ sequencing technologies to microbial genetics. Nat Rev Microbiol. 2009;7:287–296. - PubMed
-
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. - PubMed
-
- Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
