

Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels
- PMID: 20142517
- PMCID: PMC2828906
- DOI: 10.1085/jgp.200910308
Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels
Abstract
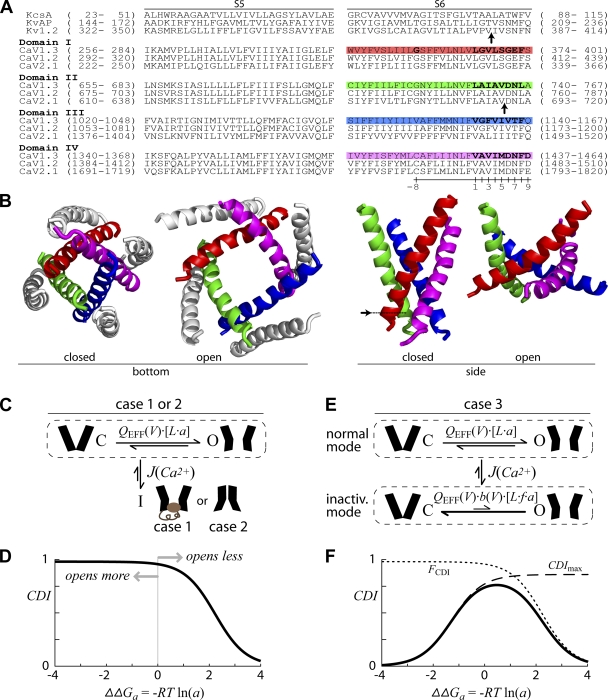
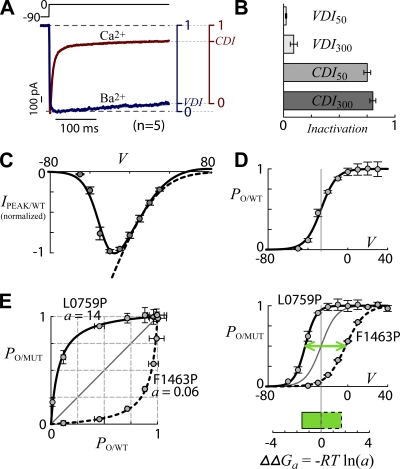
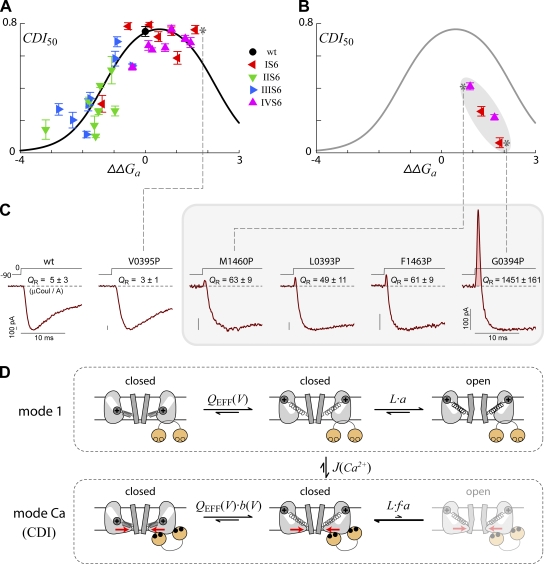
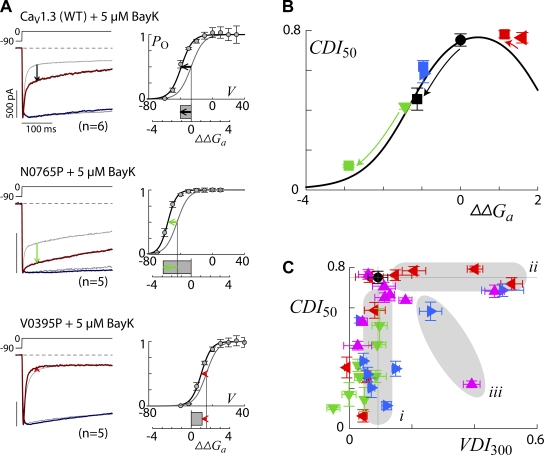
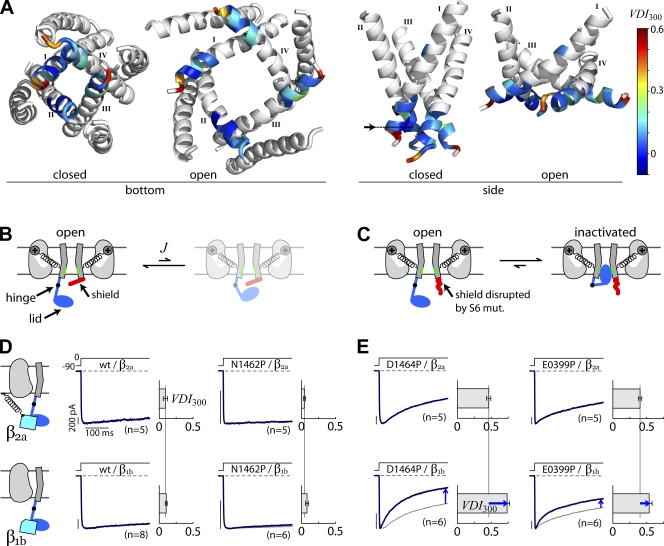
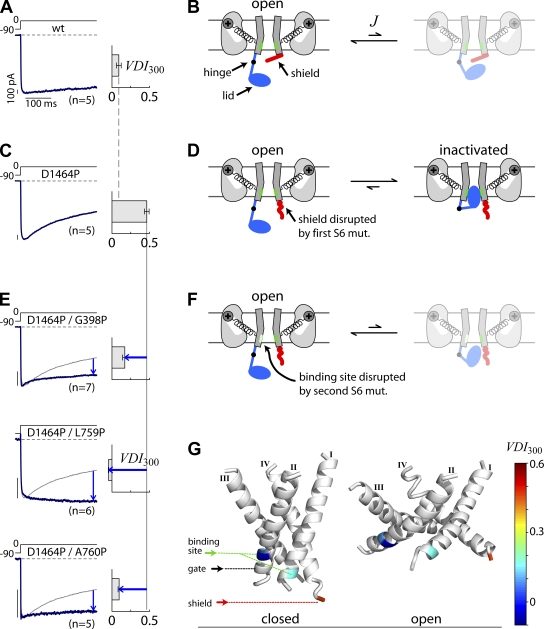
Ca(2+)/calmodulin- and voltage-dependent inactivation (CDI and VDI) comprise vital prototypes of Ca(2+) channel modulation, rich with biological consequences. Although the events initiating CDI and VDI are known, their downstream mechanisms have eluded consensus. Competing proposals include hinged-lid occlusion of channels, selectivity filter collapse, and allosteric inhibition of the activation gate. Here, novel theory predicts that perturbations of channel activation should alter inactivation in distinctive ways, depending on which hypothesis holds true. Thus, we systematically mutate the activation gate, formed by all S6 segments within Ca(V)1.3. These channels feature robust baseline CDI, and the resulting mutant library exhibits significant diversity of activation, CDI, and VDI. For CDI, a clear and previously unreported pattern emerges: activation-enhancing mutations proportionately weaken inactivation. This outcome substantiates an allosteric CDI mechanism. For VDI, the data implicate a "hinged lid-shield" mechanism, similar to a hinged-lid process, with a previously unrecognized feature. Namely, we detect a "shield" in Ca(V)1.3 channels that is specialized to repel lid closure. These findings reveal long-sought downstream mechanisms of inactivation and may furnish a framework for the understanding of Ca(2+) channelopathies involving S6 mutations.
Figures

Comment in
-
Reduction of CaV channel activities by Ca2+-CaM: inactivation or deactivation?J Gen Physiol. 2010 Apr;135(4):297-301. doi: 10.1085/jgp.201010421. Epub 2010 Mar 1. J Gen Physiol. 2010. PMID: 20194603 Free PMC article. No abstract available.
References
-
- Adams P.J., Garcia E., Snutch T.P., Spacey S.D.. 2007. Splice variant composition of P/Q-type calcium channels affects both biophysical properties and sensitivity to an FHM point mutation. Biophys. J. Abstr 2869:602A.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
