

Dynamic regulation of barrier integrity of the corneal endothelium
- PMID: 20142793
- PMCID: PMC2868144
- DOI: 10.1097/OPX.0b013e3181d39464
Dynamic regulation of barrier integrity of the corneal endothelium
Abstract
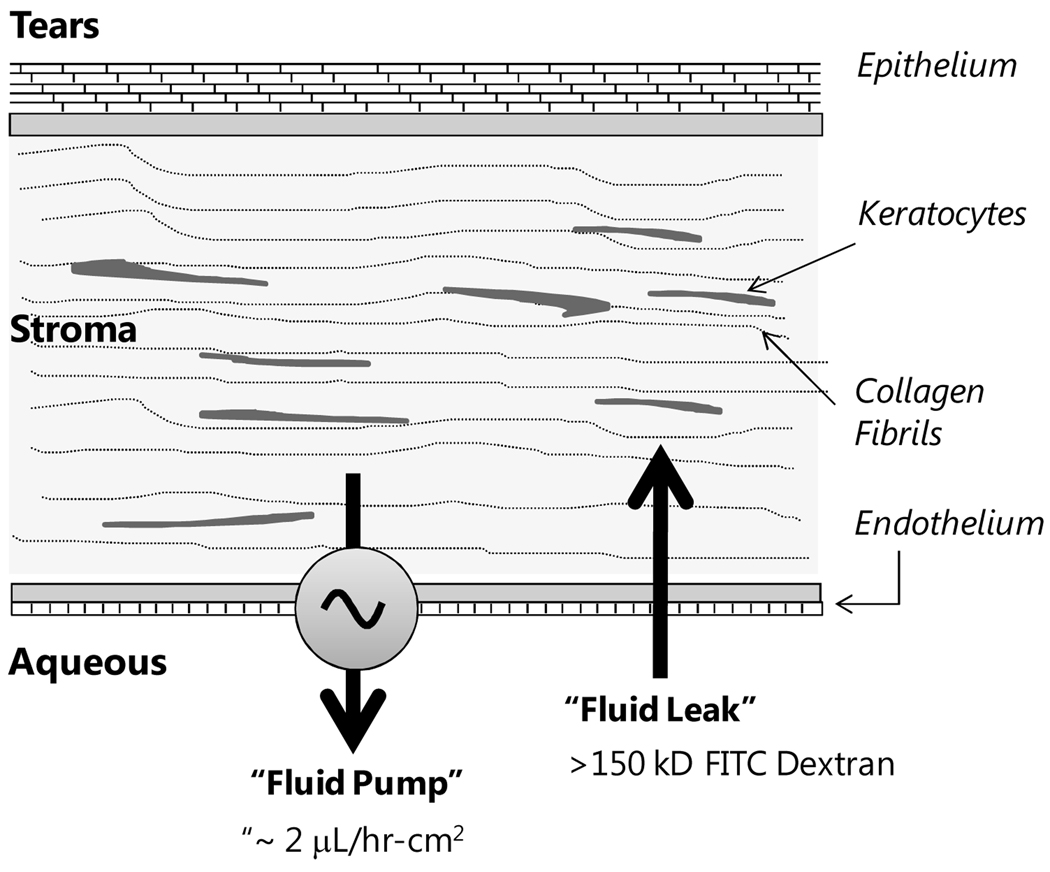
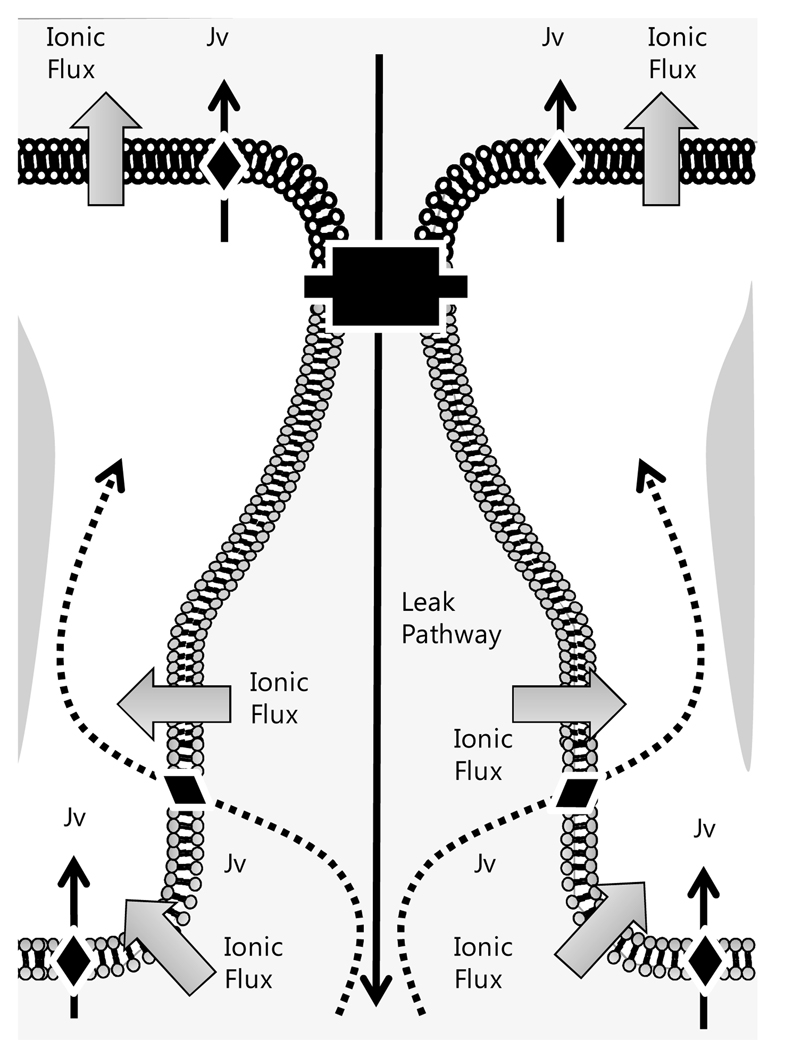
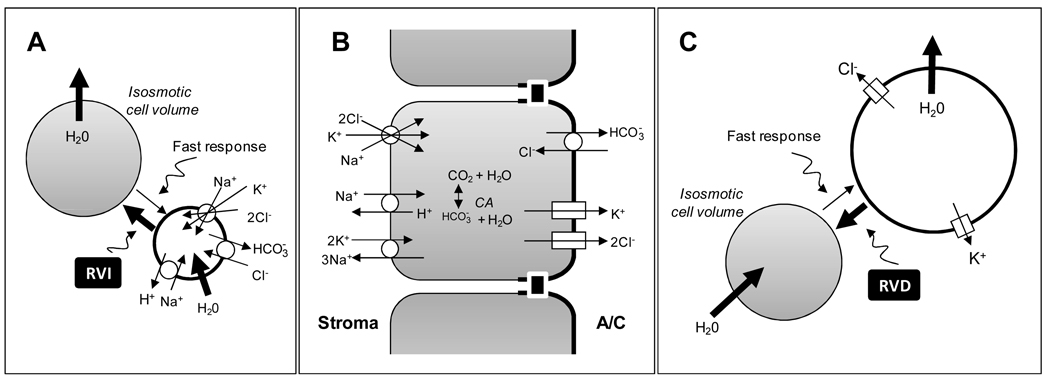
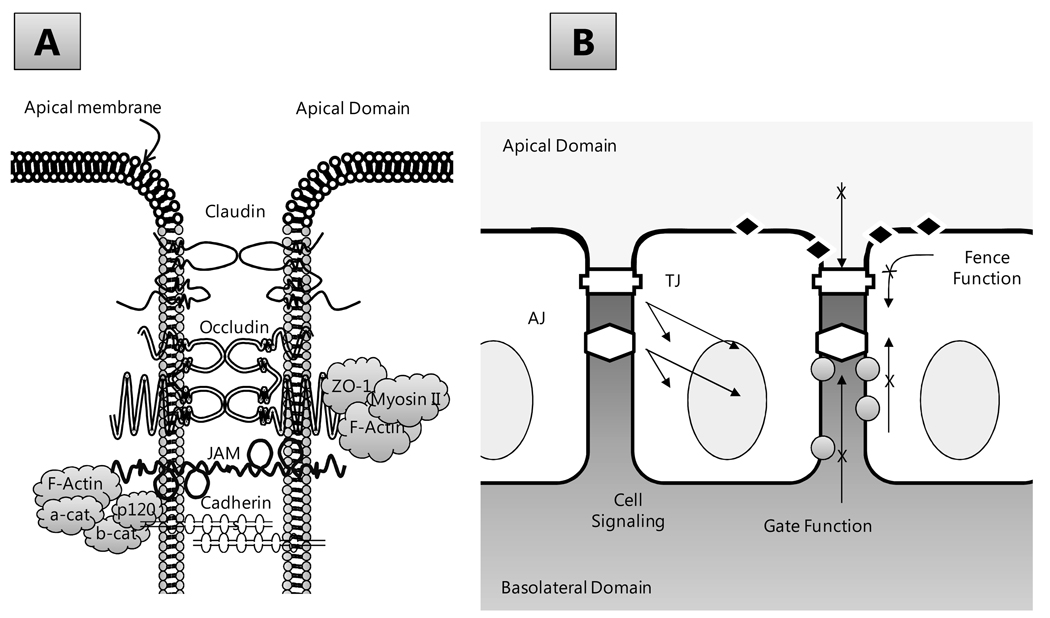
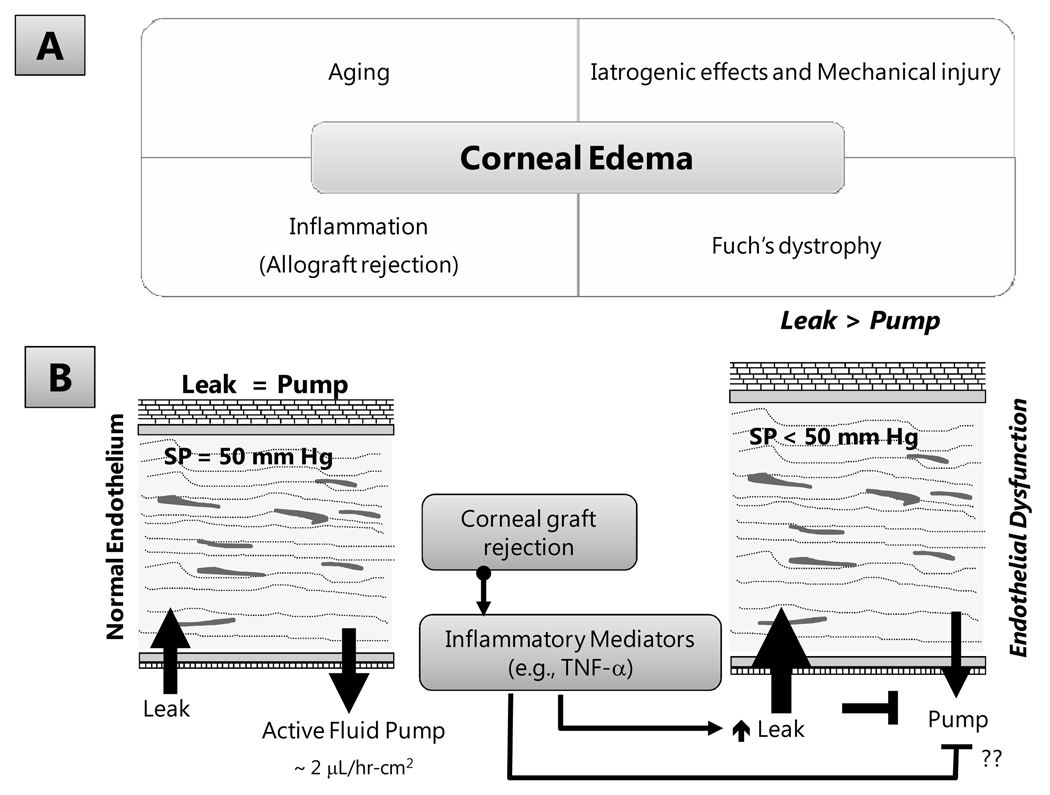
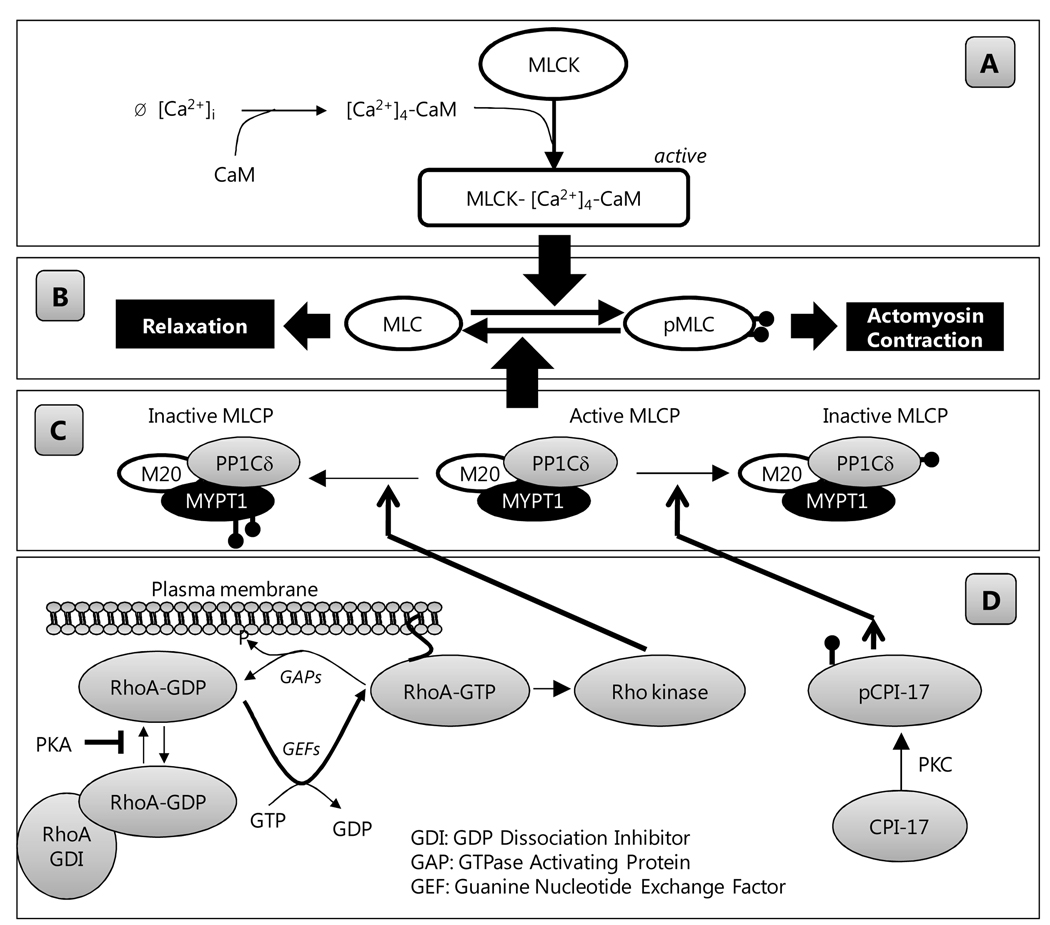
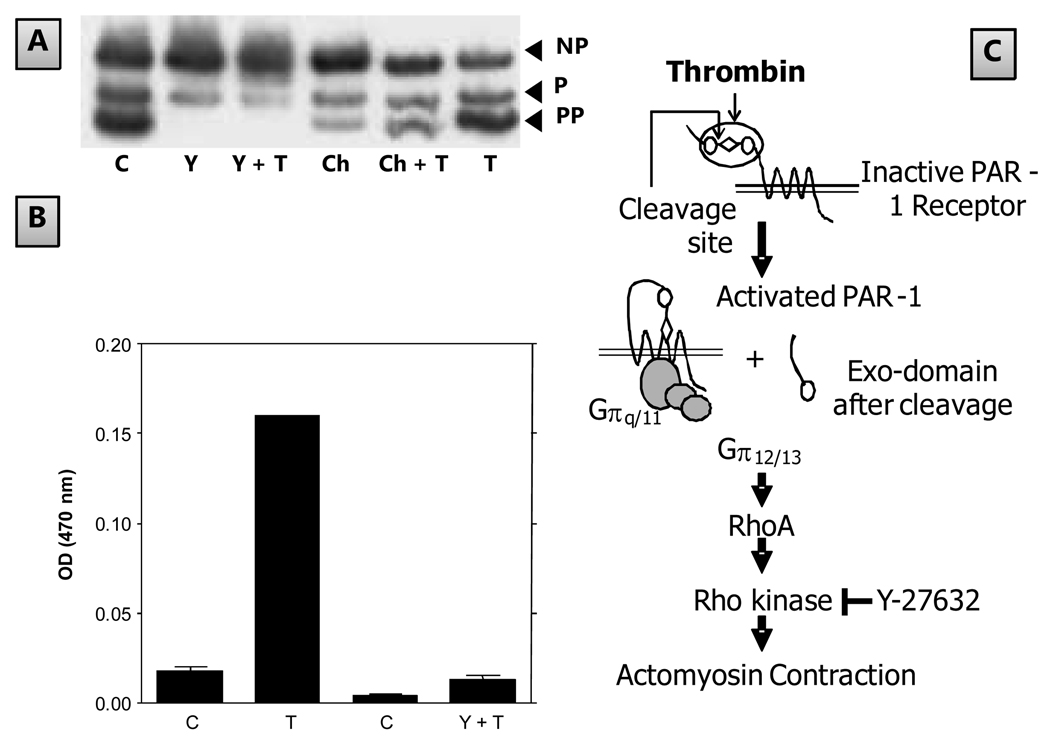
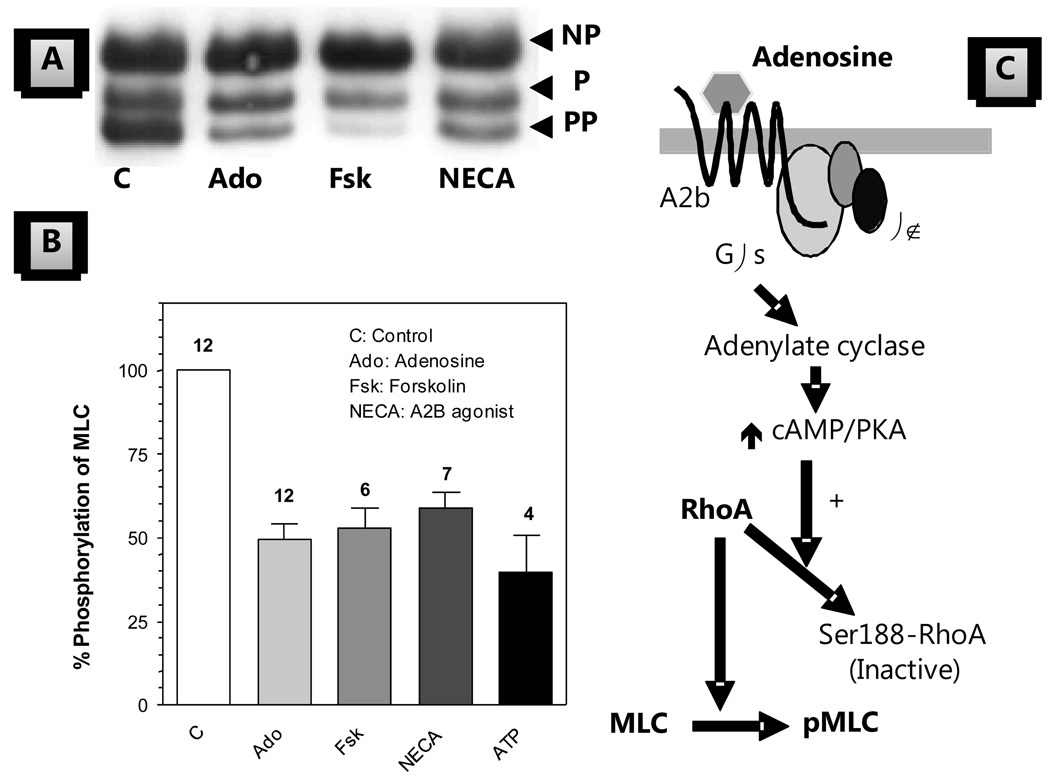
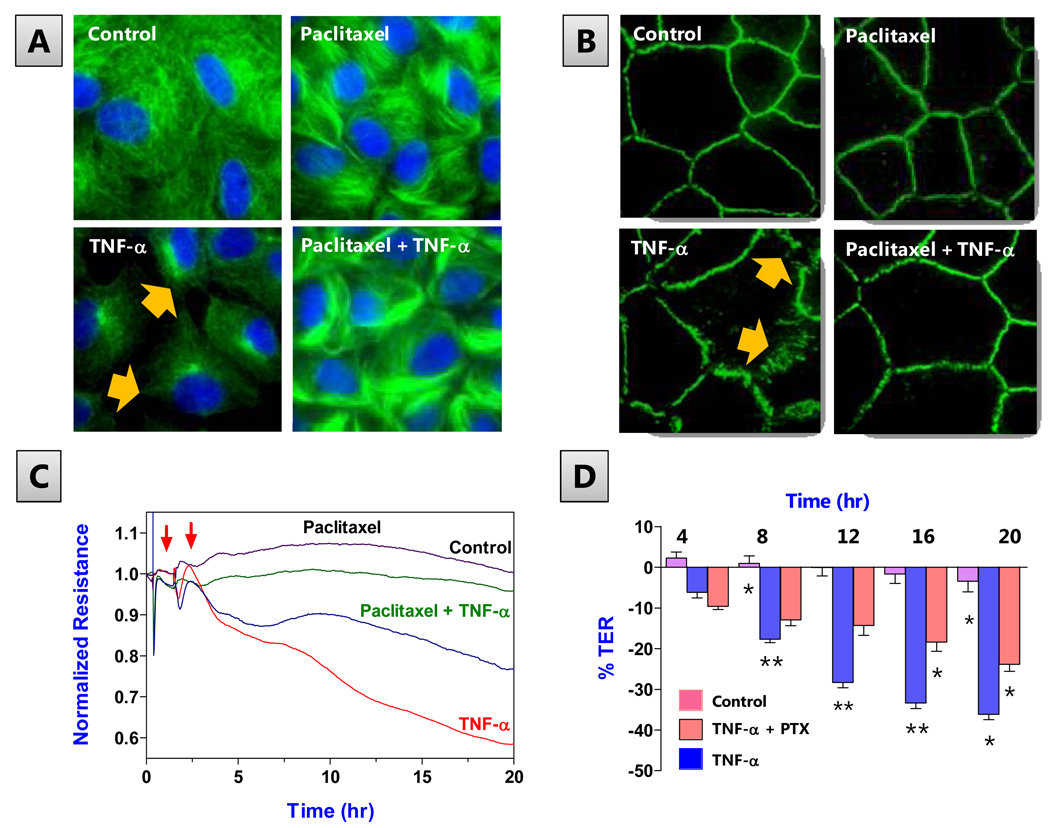
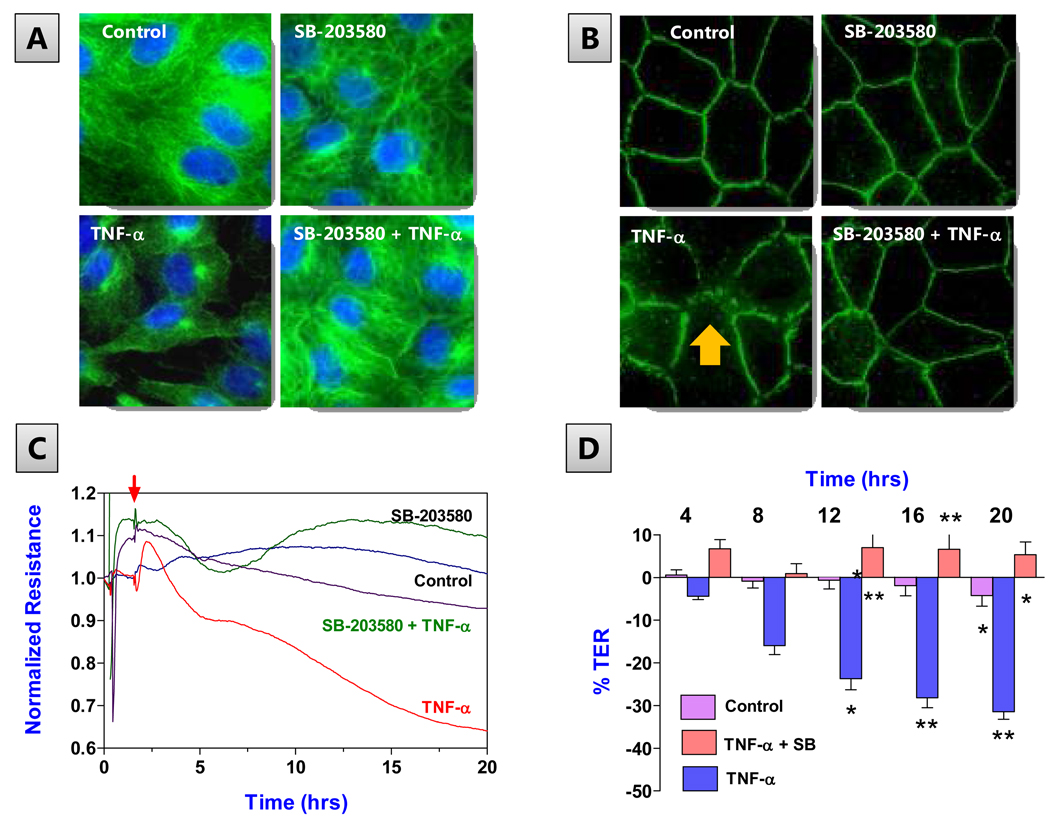
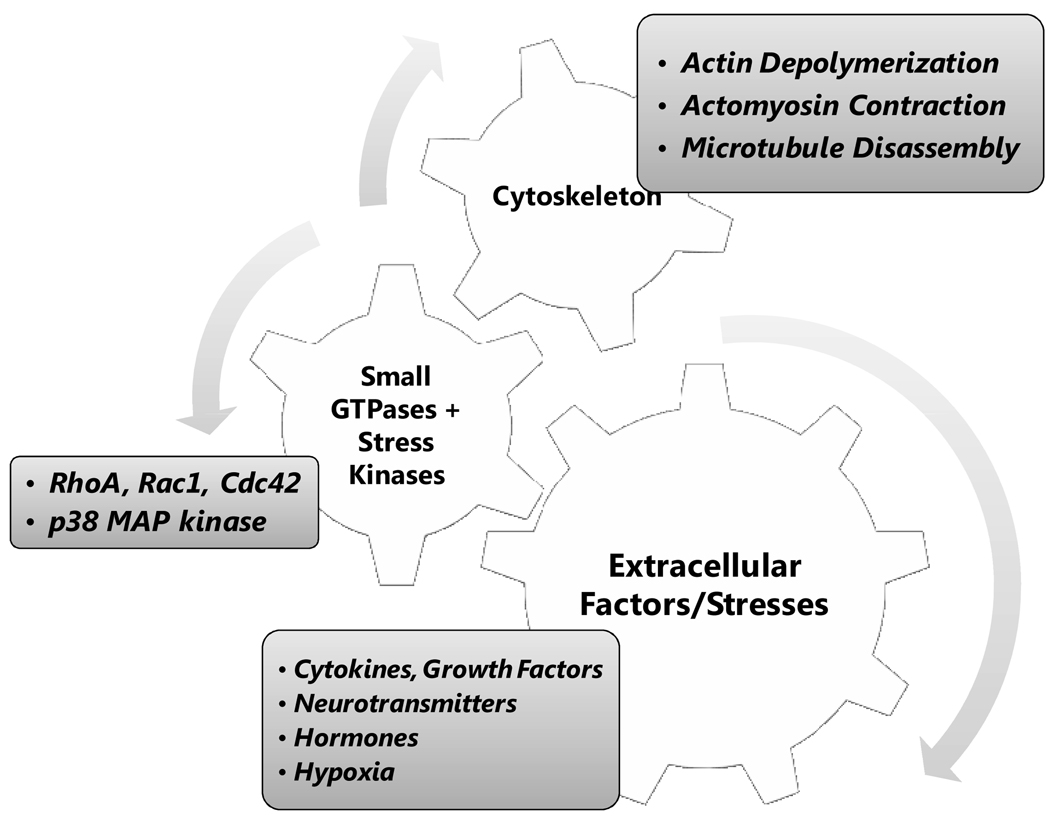
The corneal endothelium maintains stromal deturgescence, which is a prerequisite for corneal transparency. The principal challenge to stromal deturgescence is the swelling pressure associated with the hydrophilic glycosaminoglycans in the stroma. This negative pressure induces fluid leak into the stroma from the anterior chamber, but the rate of leak is restrained by the tight junctions of the endothelium. This role of the endothelium represents its barrier function. In healthy cornea, the fluid leak is counterbalanced by an active fluid pump mechanism associated with the endothelium itself. Although this pump-leak hypothesis was postulated several decades ago, the mechanisms underlying regulation of the balance between the pump and leak functions remain largely unknown. In the last couple of decades, the ion transport systems that support the fluid pump activity have been discovered. In contrast, despite significant evidence for corneal edema secondary to endothelial barrier dysfunction, the molecular aspects underlying its regulation are relatively unknown. Recent findings in our laboratory, however, indicate that barrier integrity (i.e., structural and functional integrity of the tight junctions) of the endothelium is sensitive to remodeling of its peri-junctional actomyosin ring, which is located at the apical junctional complex. This review provides a focused perspective on dynamic regulation of the barrier integrity of endothelium vis-à-vis plasticity of the peri-junctional actomyosin ring and its association with cell signaling downstream of small GTPases of the Rho family. Based on findings to date, it appears that development of specific pharmacological strategies to treat corneal edema in response to inflammatory stress would be possible in the near future.
Figures
Similar articles
-
Cell signaling in regulation of the barrier integrity of the corneal endothelium.Exp Eye Res. 2012 Feb;95(1):8-15. doi: 10.1016/j.exer.2011.09.009. Epub 2011 Sep 24. Exp Eye Res. 2012. PMID: 21963716 Free PMC article. Review.
-
12. Endothelial pump and barrier function.Exp Eye Res. 2020 Sep;198:108068. doi: 10.1016/j.exer.2020.108068. Epub 2020 Jul 11. Exp Eye Res. 2020. PMID: 32663497 Review.
-
Identity and regulation of ion transport mechanisms in the corneal endothelium.Prog Retin Eye Res. 2003 Jan;22(1):69-94. doi: 10.1016/s1350-9462(02)00059-9. Prog Retin Eye Res. 2003. PMID: 12597924 Review.
-
Evidence for a direct effect of bicarbonate on the rabbit corneal stroma.Optom Vis Sci. 1991 Sep;68(9):687-98. doi: 10.1097/00006324-199109000-00003. Optom Vis Sci. 1991. PMID: 1660589 Review.
-
Modulation of tight junction properties relevant to fluid transport across rabbit corneal endothelium.Exp Eye Res. 2007 Apr;84(4):790-8. doi: 10.1016/j.exer.2006.12.018. Epub 2007 Jan 9. Exp Eye Res. 2007. PMID: 17320078 Free PMC article.
Cited by
-
Utilizing an Ex Vivo Skin Penetration Analysis Model for Predicting Ocular Drug Penetration: A Feasibility Study with Curcumin Formulations.Pharmaceutics. 2024 Oct 6;16(10):1302. doi: 10.3390/pharmaceutics16101302. Pharmaceutics. 2024. PMID: 39458631 Free PMC article.
-
Penetration of fluorescein across the rabbit cornea from the endothelial surface.Pharm Res. 2012 Dec;29(12):3325-34. doi: 10.1007/s11095-012-0824-3. Epub 2012 Jul 20. Pharm Res. 2012. PMID: 22814903
-
Recovery of Corneal Endothelial Cells from Periphery after Injury.PLoS One. 2015 Sep 17;10(9):e0138076. doi: 10.1371/journal.pone.0138076. eCollection 2015. PLoS One. 2015. PMID: 26378928 Free PMC article.
-
SIRT1 Activation Suppresses Corneal Endothelial-Mesenchymal Transition via the TGF-β/Smad2/3 Pathway.Curr Issues Mol Biol. 2024 Dec 6;46(12):13846-13859. doi: 10.3390/cimb46120827. Curr Issues Mol Biol. 2024. PMID: 39727955 Free PMC article.
-
Current and Future Cornea Chip Models for Advancing Ophthalmic Research and Therapeutics.Adv Biol (Weinh). 2025 Jul;9(7):e2400571. doi: 10.1002/adbi.202400571. Epub 2025 Feb 17. Adv Biol (Weinh). 2025. PMID: 39962012 Free PMC article. Review.
References
-
- Edelhauser HF. The balance between corneal transparency and edema: the Proctor Lecture. Invest Ophthalmol Vis Sci. 2006;47:1754–1767. - PubMed
-
- Riley M. Pump and leak in regulation of fluid transport in rabbit cornea. Curr Eye Res. 1985;4:371–376. - PubMed
-
- Fischbarg J, Maurice DM. An update on corneal hydration control. Exp Eye Res. 2004;78:537–541. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
