

A constructive approach for discovering new drug leads: Using a kernel methodology for the inverse-QSAR problem
- PMID: 20142987
- PMCID: PMC2816860
- DOI: 10.1186/1758-2946-1-4
A constructive approach for discovering new drug leads: Using a kernel methodology for the inverse-QSAR problem
Abstract
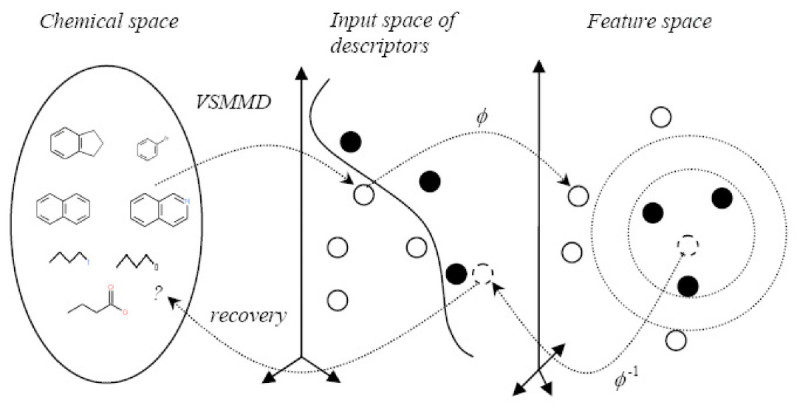
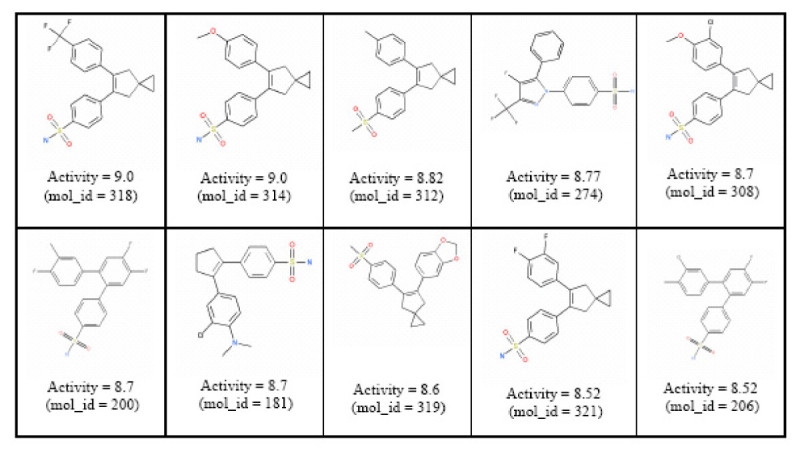
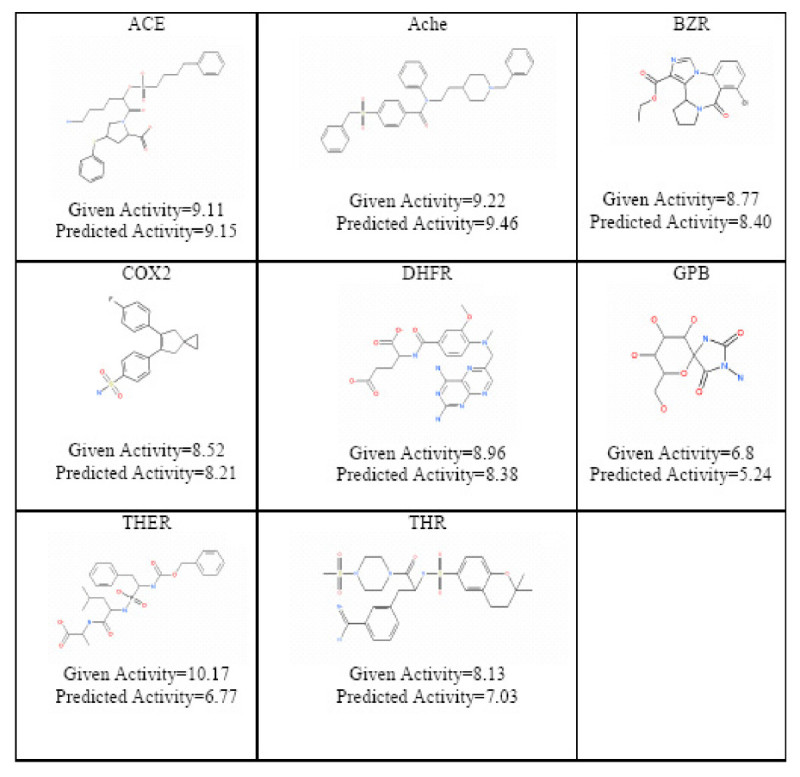
Background: The inverse-QSAR problem seeks to find a new molecular descriptor from which one can recover the structure of a molecule that possess a desired activity or property. Surprisingly, there are very few papers providing solutions to this problem. It is a difficult problem because the molecular descriptors involved with the inverse-QSAR algorithm must adequately address the forward QSAR problem for a given biological activity if the subsequent recovery phase is to be meaningful. In addition, one should be able to construct a feasible molecule from such a descriptor. The difficulty of recovering the molecule from its descriptor is the major limitation of most inverse-QSAR methods.
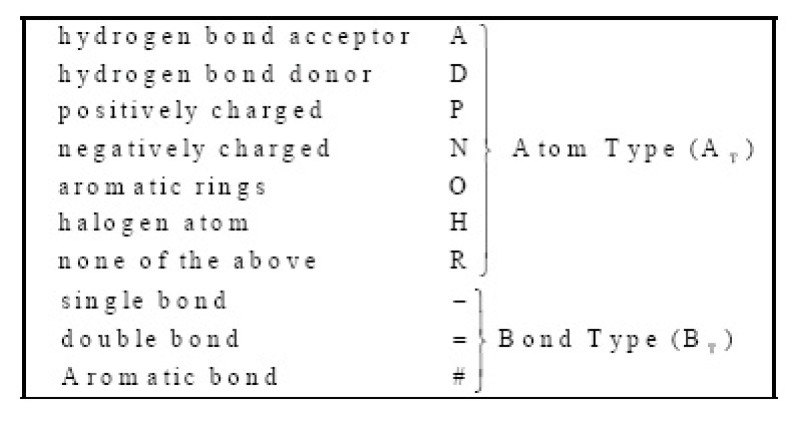
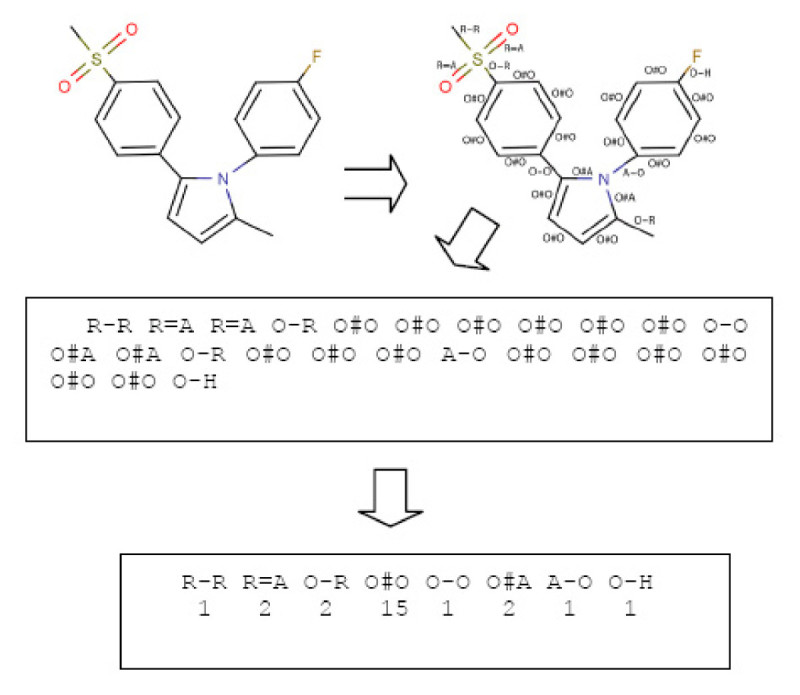
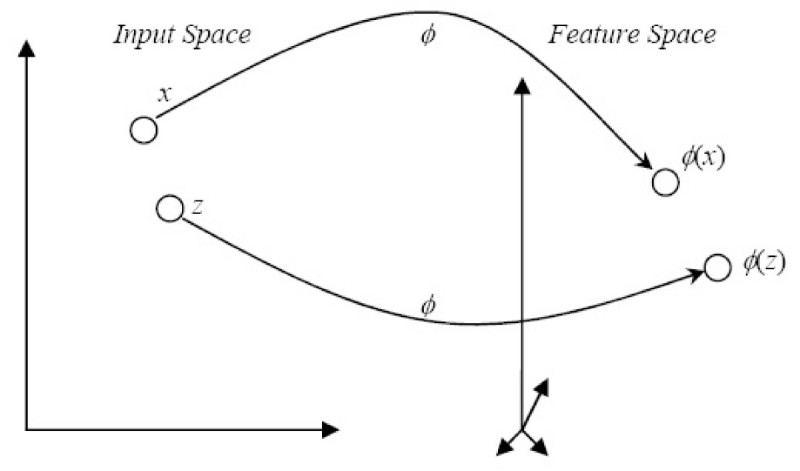
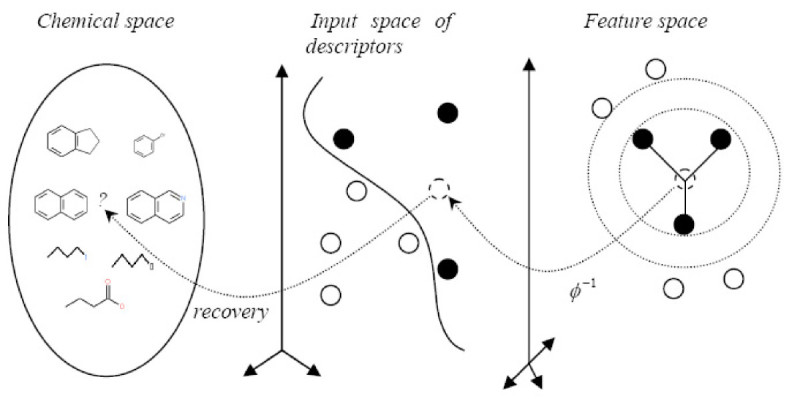
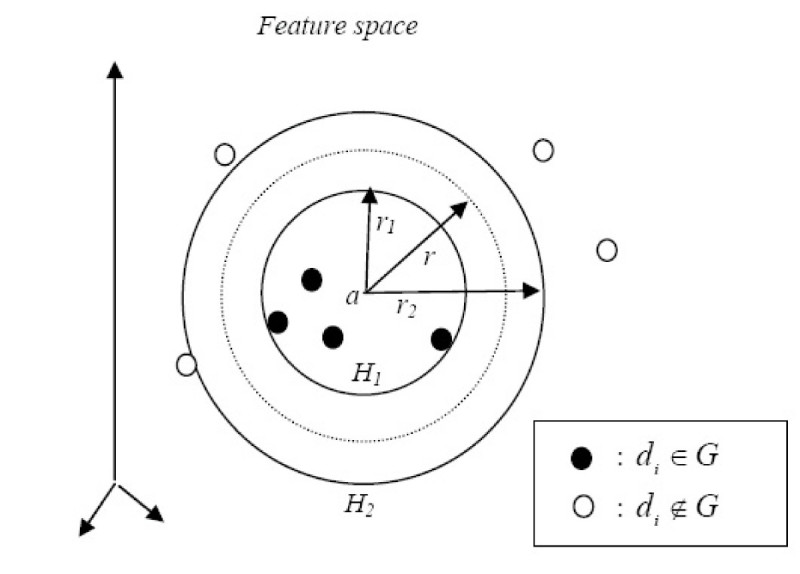
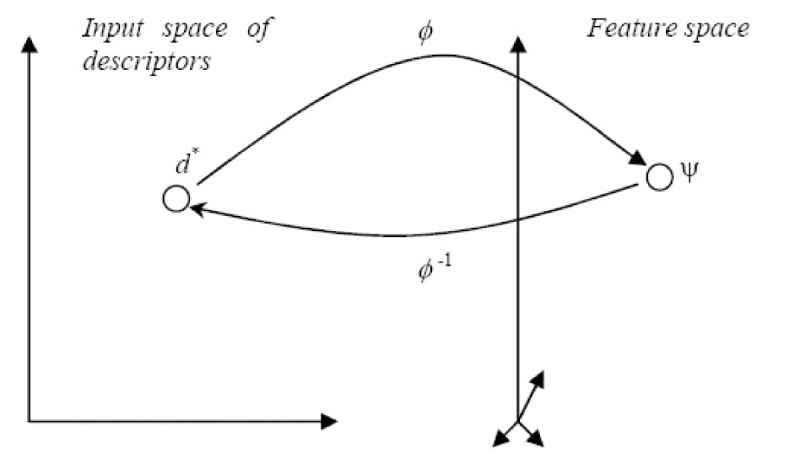
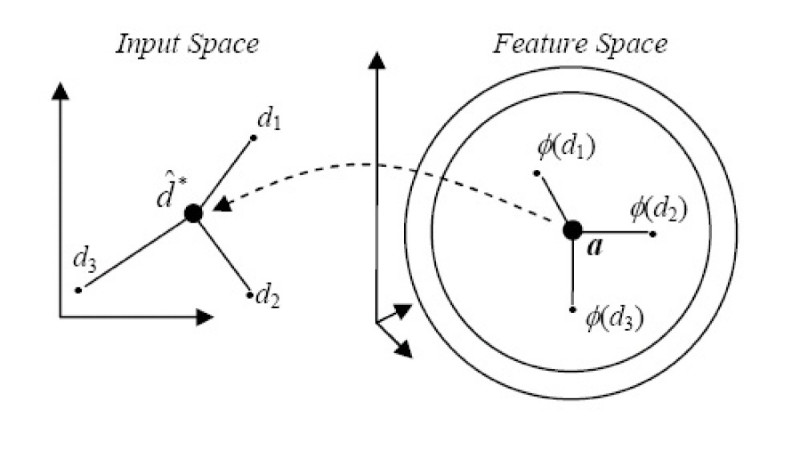
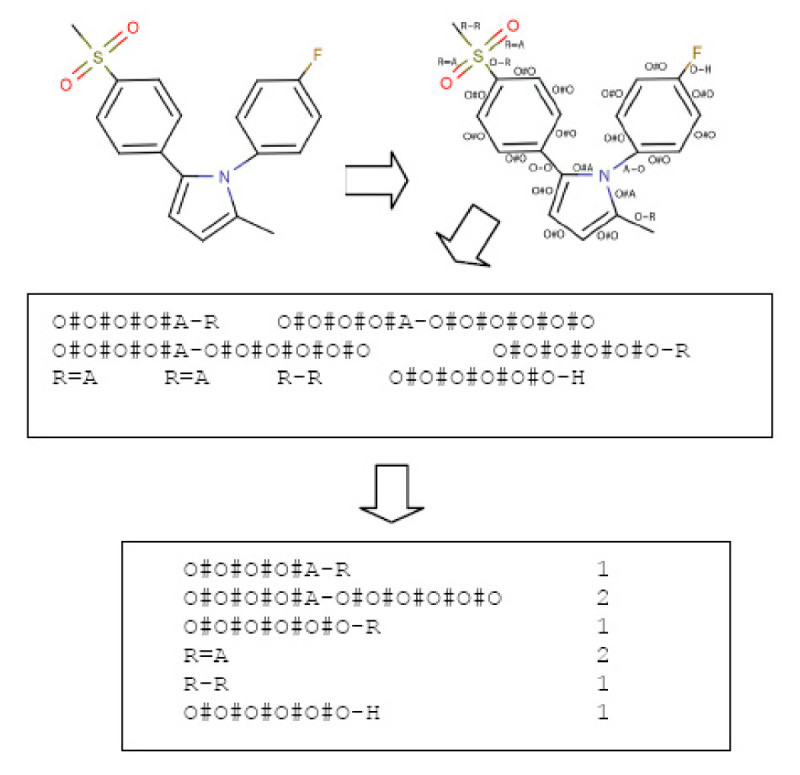
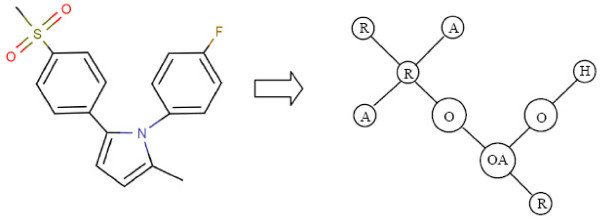
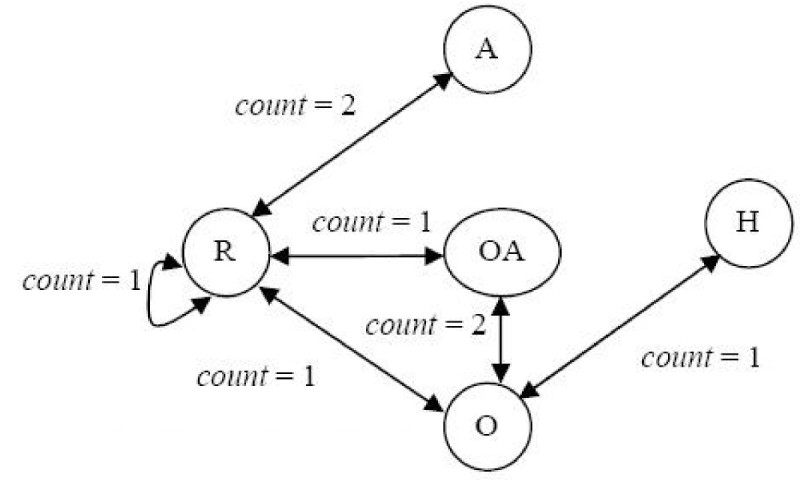
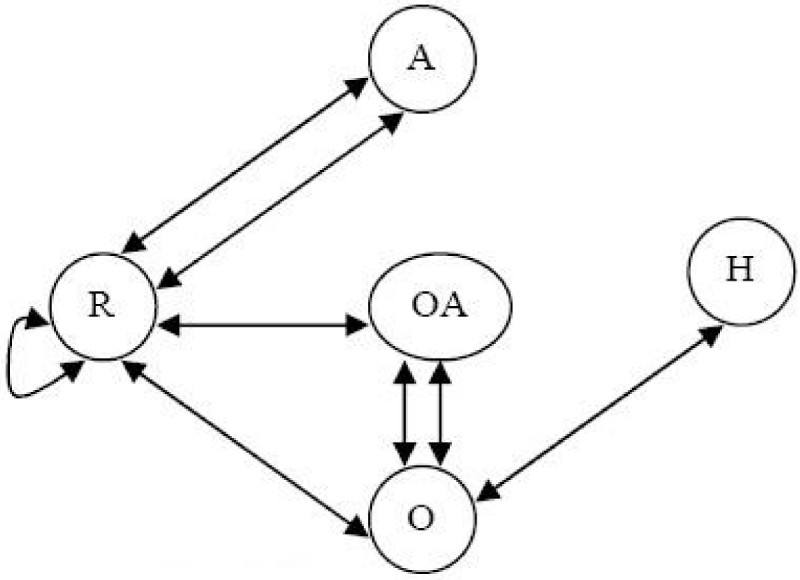
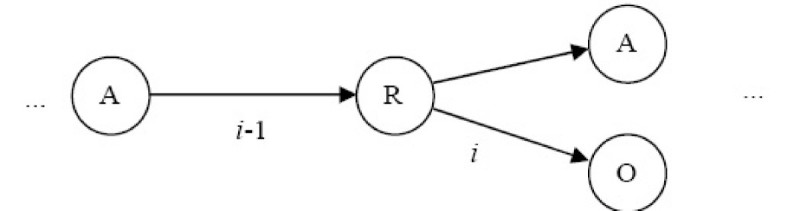
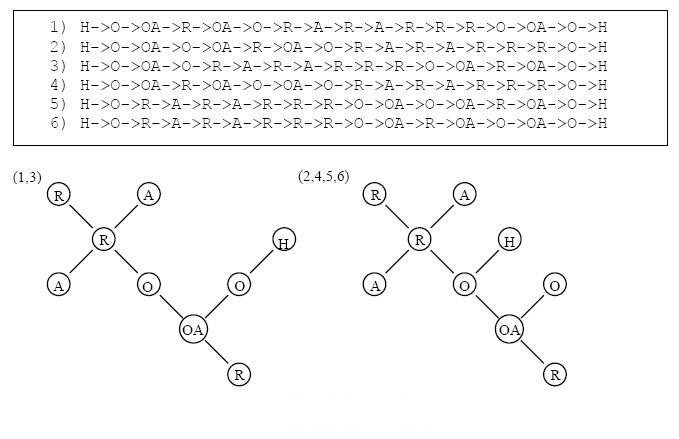
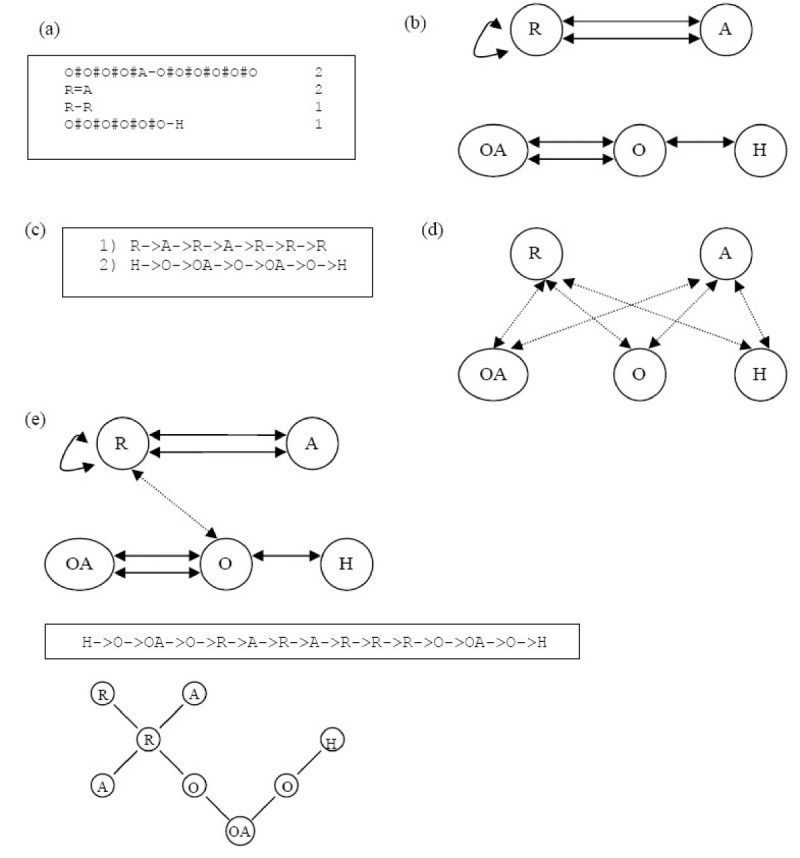
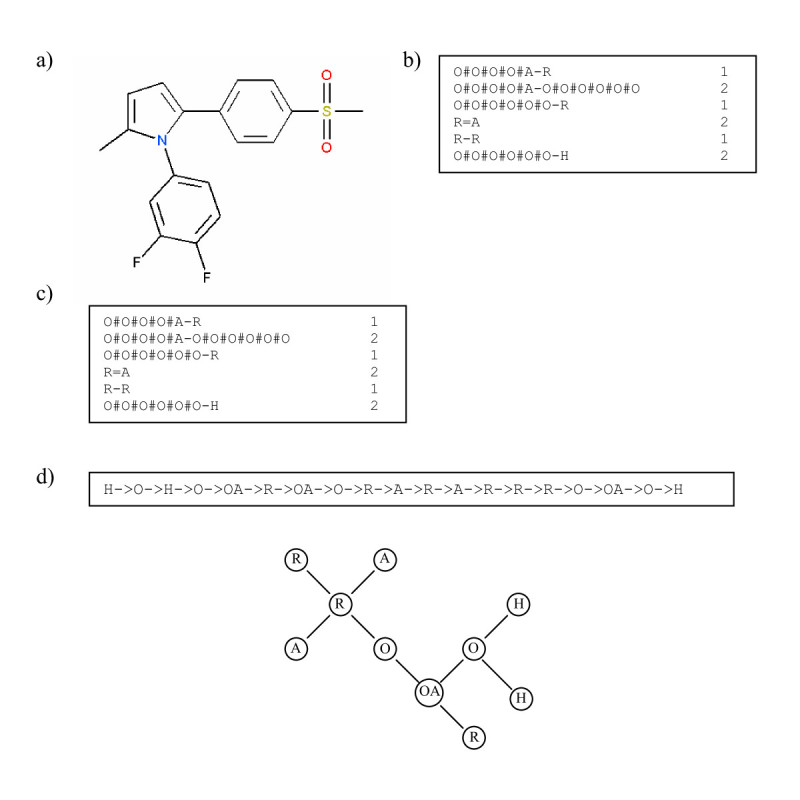
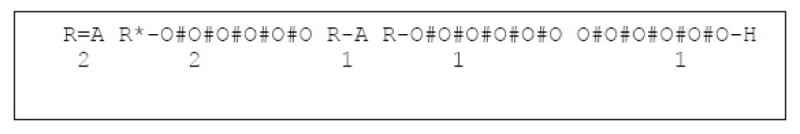
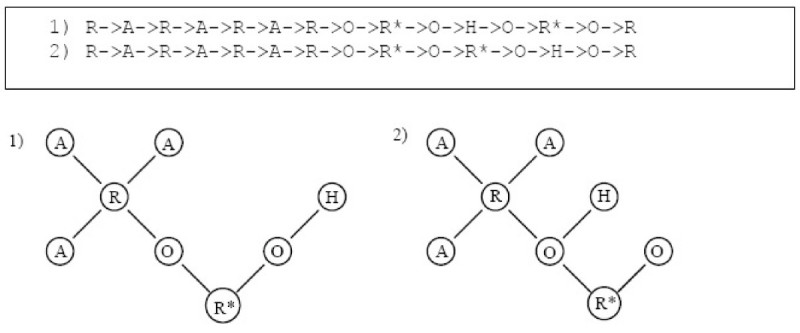
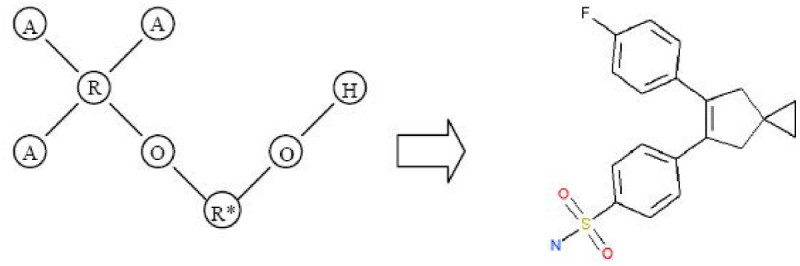
Results: In this paper, we describe the reversibility of our previously reported descriptor, the vector space model molecular descriptor (VSMMD) based on a vector space model that is suitable for kernel studies in QSAR modeling. Our inverse-QSAR approach can be described using five steps: (1) generate the VSMMD for the compounds in the training set; (2) map the VSMMD in the input space to the kernel feature space using an appropriate kernel function; (3) design or generate a new point in the kernel feature space using a kernel feature space algorithm; (4) map the feature space point back to the input space of descriptors using a pre-image approximation algorithm; (5) build the molecular structure template using our VSMMD molecule recovery algorithm.
Conclusion: The empirical results reported in this paper show that our strategy of using kernel methodology for an inverse-Quantitative Structure-Activity Relationship is sufficiently powerful to find a meaningful solution for practical problems.
Figures

References
-
- Sharp KA. Potential functions for virtual screening and ligand binding calculations: Some theoretical considerations . In: Alvarez J, Shoichet B, editors. Virtual Screening in Drug Discovery. New York: Taylor & Francis; 2005. pp. 229–248.
-
- Todeschini R, Consonni V. Handbook of molecular descriptors. Weinheim: Wiley-VCH; 2000.
LinkOut - more resources
Full Text Sources
Other Literature Sources
