

Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes
- PMID: 20145242
- PMCID: PMC2857037
- DOI: 10.1074/jbc.M109.076984
Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes
Abstract
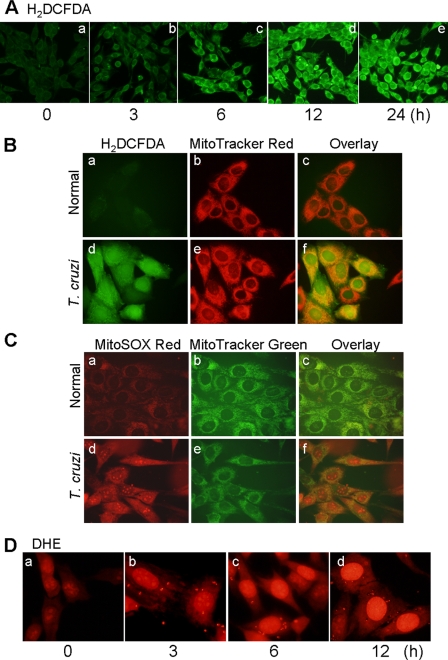
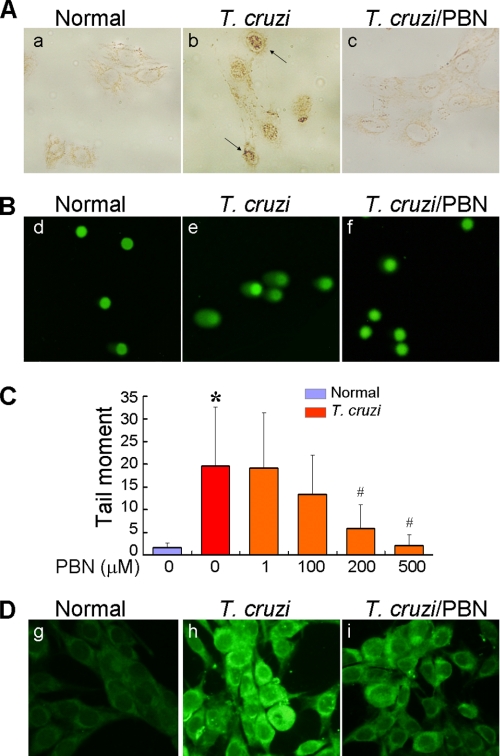
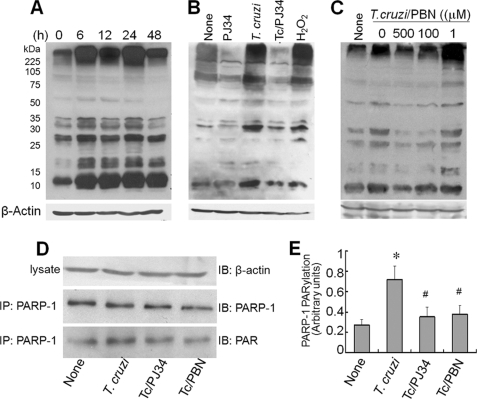
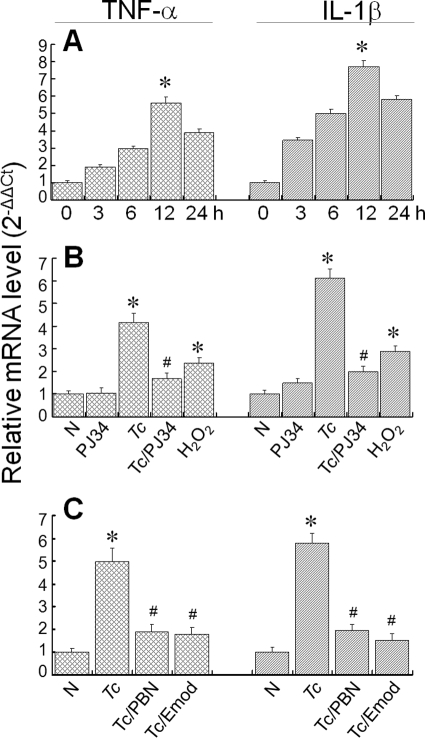
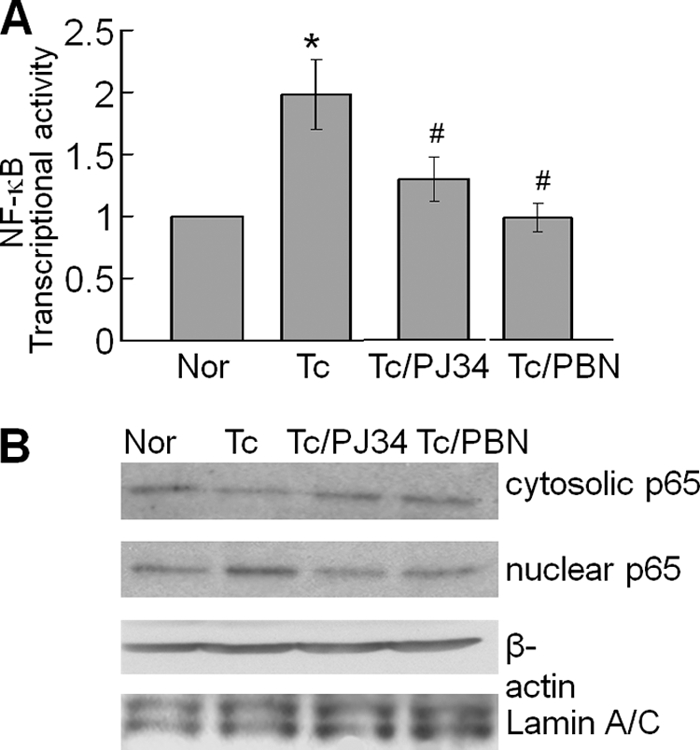
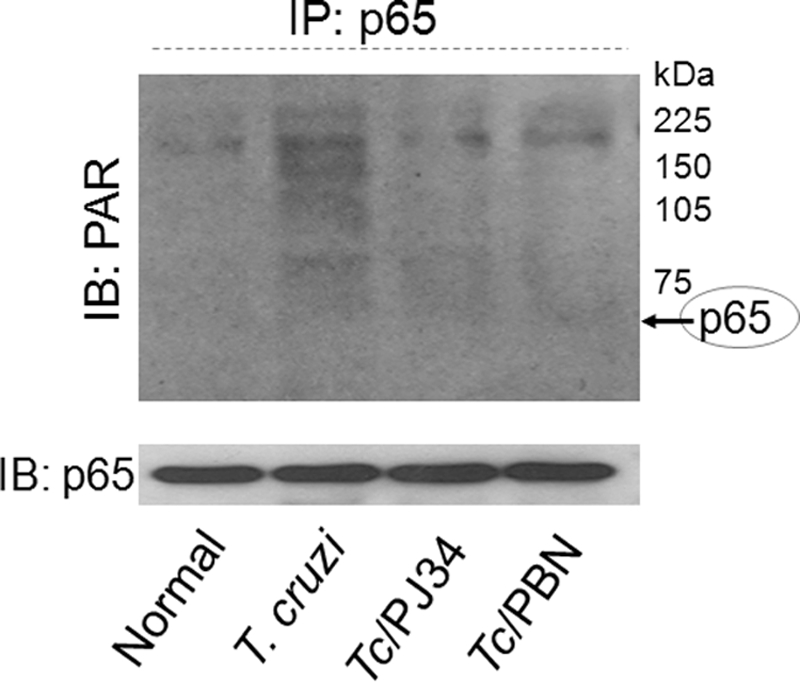
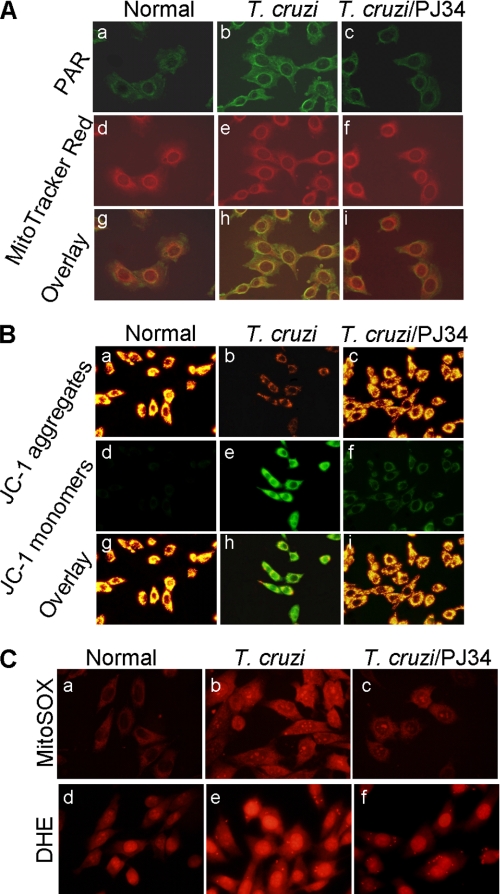
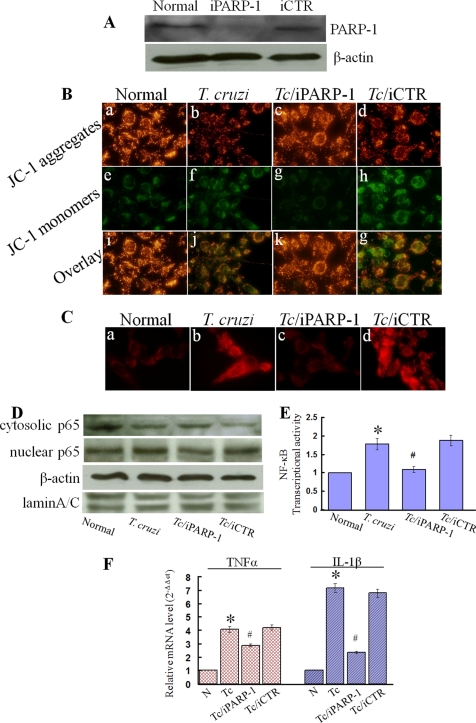
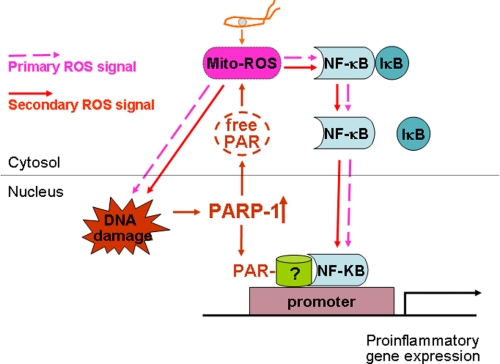
In this study, we demonstrate that human cardiomyocytes (AC16) produce reactive oxygen species (ROS) and inflammatory cytokines in response to Trypanosoma cruzi. ROS were primarily produced by mitochondria, some of which diffused to cytosol of infected cardiomyocytes. These ROS resulted in an increase in 8-hydroxyguanine lesions and DNA fragmentation that signaled PARP-1 activation evidenced by poly(ADP-ribose) (PAR) modification of PARP-1 and other proteins in infected cardiomyocytes. Phenyl-alpha-tert-butylnitrone blocked the mitochondrial ROS (mtROS) formation, DNA damage, and PARP-1 activation in infected cardiomyocytes. Further inhibition studies demonstrated that ROS and PARP-1 signaled TNF-alpha and IL-1beta expression in infected cardiomyocytes. ROS directly signaled the nuclear translocation of RelA (p65), NF-kappaB activation, and cytokine gene expression. PARP-1 exhibited no direct interaction with p65 and did not signal its translocation to nuclei in infected cardiomyocytes. Instead, PARP-1 contributed to PAR modification of p65-interacting nuclear proteins and assembly of the NF-kappaB transcription complex. PJ34 (PARP-1 inhibitor) also prevented mitochondrial poly(ADP-ribosyl)ation (PARylation) and ROS formation. We conclude that T. cruzi-mediated mtROS provide primary stimulus for PARP-1-NF-kappaB activation and cytokine gene expression in infected cardiomyocytes. PAR modification of mitochondrial membranes then results in a feedback cycle of mtROS formation and DNA damage/PARP-1 activation. ROS, either through direct modulation of cytosolic NF-kappaB, or via PARP-1-dependent PAR modification of p65-interacting nuclear proteins, contributes to cytokine gene expression. Our results demonstrate a link between ROS and inflammatory responses in cardiomyocytes infected by T. cruzi and provide a clue to the pathomechanism of sustained inflammation in Chagas disease.
Figures
Similar articles
-
Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation.J Immunol. 2010 Aug 1;185(3):1894-902. doi: 10.4049/jimmunol.1000646. Epub 2010 Jul 7. J Immunol. 2010. PMID: 20610652 Free PMC article.
-
Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-κB transcription in murine macrophages.Hum Immunol. 2012 May;73(5):439-47. doi: 10.1016/j.humimm.2012.02.002. Epub 2012 Feb 21. Hum Immunol. 2012. PMID: 22391342
-
Critical role of the automodification of poly(ADP-ribose) polymerase-1 in nuclear factor-kappaB-dependent gene expression in primary cultured mouse glial cells.J Biol Chem. 2004 Oct 8;279(41):42774-86. doi: 10.1074/jbc.M407923200. Epub 2004 Aug 9. J Biol Chem. 2004. PMID: 15302869
-
The poly(ADP-ribose) polymerases (PARPs): new roles in intracellular transport.Cell Signal. 2012 Jan;24(1):1-8. doi: 10.1016/j.cellsig.2011.07.019. Epub 2011 Aug 5. Cell Signal. 2012. PMID: 21840394 Review.
-
Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders.Cardiovasc Toxicol. 2018 Dec;18(6):493-506. doi: 10.1007/s12012-018-9462-2. Cardiovasc Toxicol. 2018. PMID: 29968072 Review.
Cited by
-
Host cell poly(ADP-ribose) glycohydrolase is crucial for Trypanosoma cruzi infection cycle.PLoS One. 2013 Jun 12;8(6):e67356. doi: 10.1371/journal.pone.0067356. Print 2013. PLoS One. 2013. PMID: 23776710 Free PMC article.
-
Trypanocidal action of (-)-elatol involves an oxidative stress triggered by mitochondria dysfunction.Mar Drugs. 2012 Aug;10(8):1631-1646. doi: 10.3390/md10081631. Epub 2012 Aug 3. Mar Drugs. 2012. PMID: 23015766 Free PMC article.
-
Epidemiology and pathogenesis of maternal-fetal transmission of Trypanosoma cruzi and a case for vaccine development against congenital Chagas disease.Biochim Biophys Acta Mol Basis Dis. 2020 Mar 1;1866(3):165591. doi: 10.1016/j.bbadis.2019.165591. Epub 2019 Oct 31. Biochim Biophys Acta Mol Basis Dis. 2020. PMID: 31678160 Free PMC article. Review.
-
The repositioned drugs disulfiram/diethyldithiocarbamate combined to benznidazole: Searching for Chagas disease selective therapy, preventing toxicity and drug resistance.Front Cell Infect Microbiol. 2022 Jul 29;12:926699. doi: 10.3389/fcimb.2022.926699. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35967878 Free PMC article.
-
Serum-mediated activation of macrophages reflects TcVac2 vaccine efficacy against Chagas disease.Infect Immun. 2014 Apr;82(4):1382-9. doi: 10.1128/IAI.01186-13. Epub 2014 Jan 13. Infect Immun. 2014. PMID: 24421046 Free PMC article.
References
-
- World Health Organization (2006) Report of the Scientific Working Group on Chagas Disease, UNDP/World Bank/WHO
-
- Talvani A., Rocha M. O., Barcelos L. S., Gomes Y. M., Ribeiro A. L., Teixeira M. M. (2004) Clin. Infect. Dis. 38, 943–950 - PubMed
-
- Rossi M. A., Ramos S. G., Bestetti R. B. (2003) Front. Biosci. 8, 94–109 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
