

New immune pathways from chronic post-viral lung disease
- PMID: 20146716
- PMCID: PMC3060024
- DOI: 10.1111/j.1749-6632.2009.05136.x
New immune pathways from chronic post-viral lung disease
Abstract
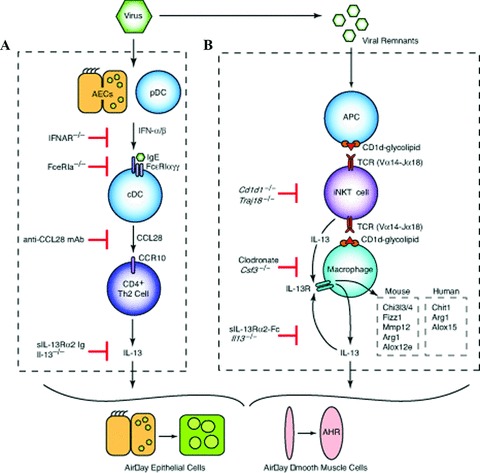
To better understand the immunopathogenesis of chronic inflammatory lung disease, we established a mouse model of disease that develops after respiratory viral infection. The disease that develops in this model is similar to chronic obstructive lung disease in humans. Using this model we have characterized two distinct phases in the chronic disease process. The first phase appears at three weeks after viral infection and depends on type I interferon-dependent expression and then subsequent activation of the high-affinity IgE receptor (FcepsilonRI) on conventional lung dendritic cells, which in turn recruit IL-13-producing CD4+ T cells to the lower airways. The second phase becomes maximal at seven weeks after infection and depends on invariant natural killer T (iNKT) cells and lung macrophages. Cellular cross-talk relies on interactions between the semi-invariant Valpha14Jalpha18 T-cell receptor on lung iNKT cells and CD1d on macrophages as well as iNKT cell-derived IL-13 and IL-13 receptor on macrophages. These interactions drive macrophages to a pattern of alternative activation and overproduction of IL-13. This innate immune axis is also activated in patients with chronic obstructive lung disease, as evidenced by increased numbers of iNKT cells and IL-13-producing alternatively activated macrophages marked by chitinase 1 production. Together the findings identify two new immune pathways responsible for early and late phases of chronic inflammatory lung disease in experimental and clinical settings. These findings extend our understanding of the complex mechanisms that underlie chronic obstructive lung disease and provide useful targets for diagnosis and therapy of this common disorder.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Manz, R.A. et al 2006. Immunological memory stabilizing autoreactivity. Curr. Top. Microbiol. Immunol. 305: 241–257. - PubMed
-
- O’Garra, A. & Murphy K.. 1993. T‐cell subsets in autoimmunity. Curr. Opin. Immunol. 5: 880–886. - PubMed
-
- Recher, M. & Lang K.S.. 2006. Innate (over)immunity and adaptive autoimmune disease. Curr. Top. Microbiol. Immunol. 305: 89–104. - PubMed
-
- Murray, C.J. & Lopez A.D.. 1997. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 349: 1498–1504. - PubMed
-
- Robinson, D. et al 1993. Activation of CD4+ T cells, increased TH2‐type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J. Allergy Clin. Immunol. 92: 313–324. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
