

Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx
- PMID: 20147399
- PMCID: PMC2849514
- DOI: 10.1128/JVI.02597-09
Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx
Abstract
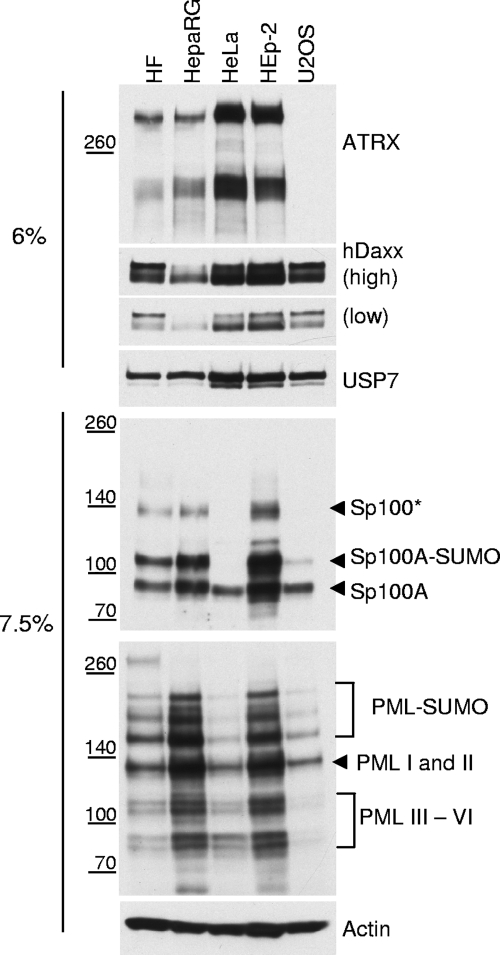
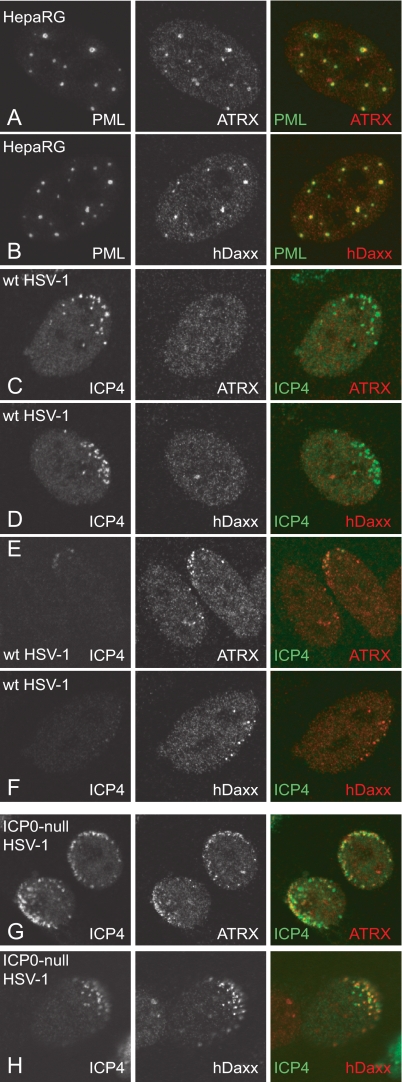
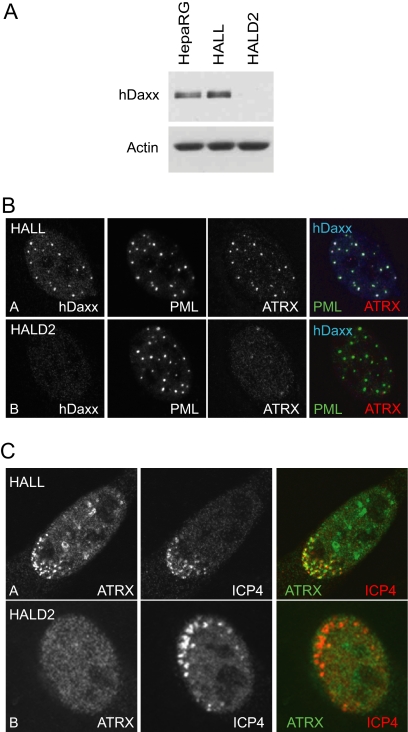
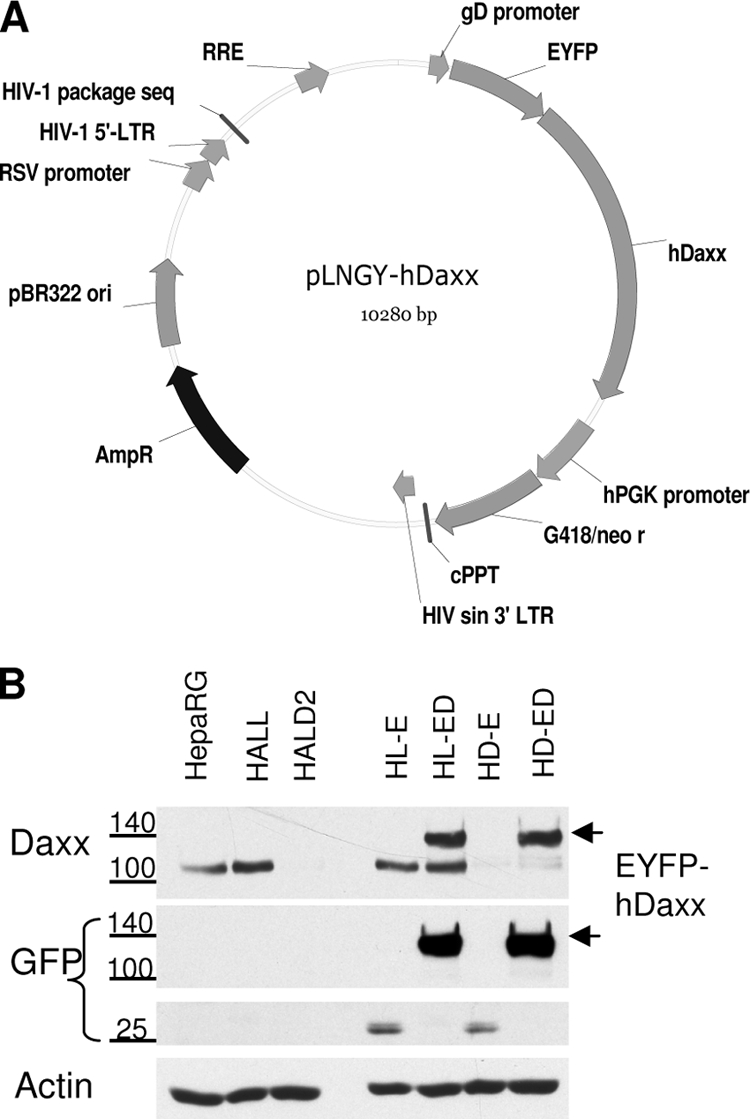
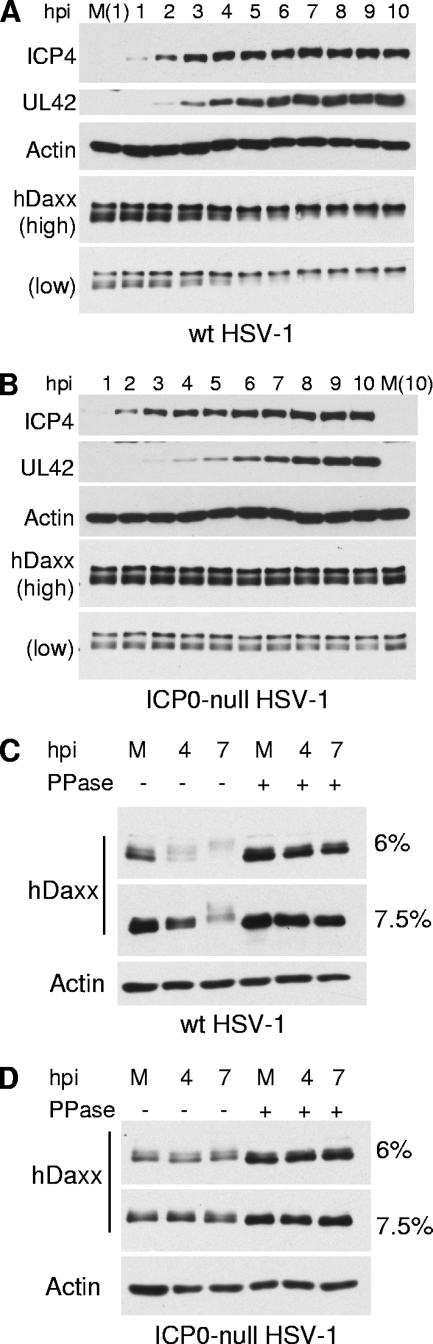
Herpes simplex virus type 1 (HSV-1) immediate-early gene product ICP0 activates lytic infection and relieves cell-mediated repression of viral gene expression. This repression is conferred by preexisting cellular proteins and is commonly referred to as intrinsic antiviral resistance or intrinsic defense. PML and Sp100, two core components of nuclear substructures known as ND10 or PML nuclear bodies, contribute to intrinsic resistance, but it is clear that other proteins must also be involved. We have tested the hypothesis that additional ND10 factors, particularly those that are involved in chromatin remodeling, may have roles in intrinsic resistance against HSV-1 infection. The two ND10 component proteins investigated in this report are ATRX and hDaxx, which are known to interact with each other and comprise components of a repressive chromatin-remodeling complex. We generated stable cell lines in which endogenous ATRX or hDaxx expression is severely suppressed by RNA interference. We found increases in both gene expression and plaque formation induced by ICP0-null mutant HSV-1 in both ATRX- and hDaxx-depleted cells. Reconstitution of wild-type hDaxx expression reversed the effects of hDaxx depletion, but reconstitution with a mutant form of hDaxx unable to interact with ATRX did not. Our results suggest that ATRX and hDaxx act as a complex that contributes to intrinsic antiviral resistance to HSV-1 infection, which is counteracted by ICP0.
Figures
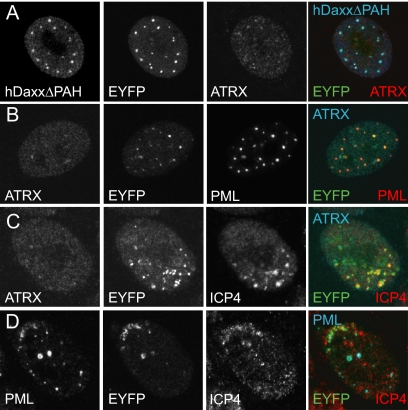
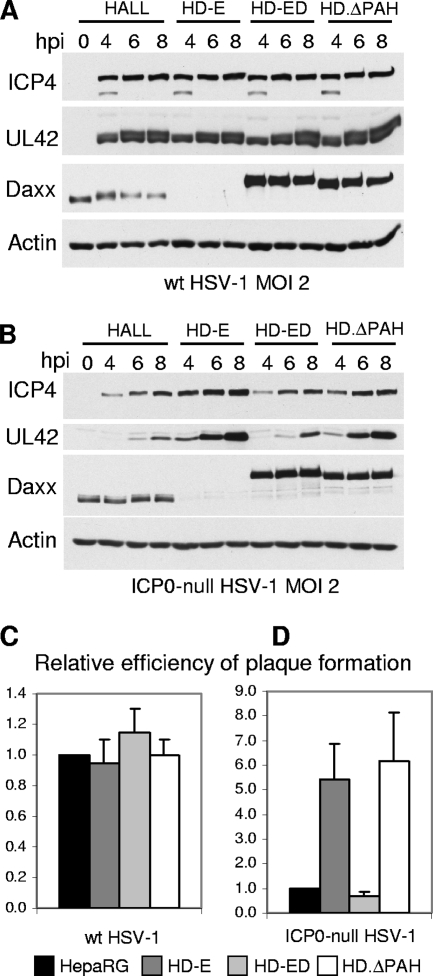
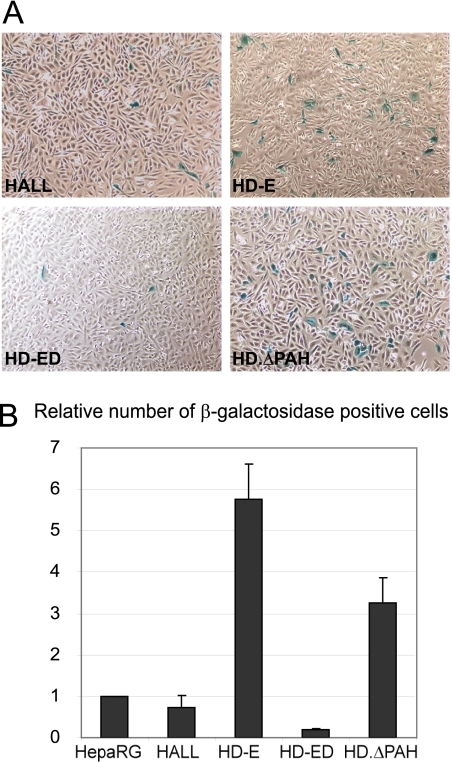
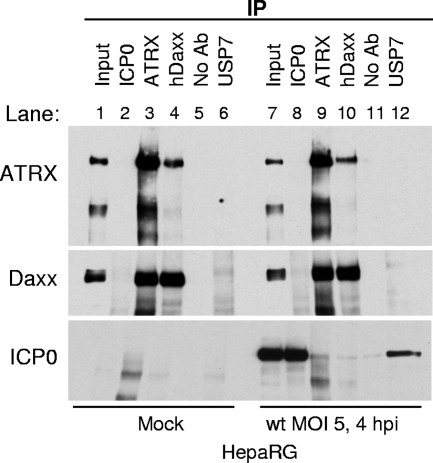
Similar articles
-
Stimulation of the Replication of ICP0-Null Mutant Herpes Simplex Virus 1 and pp71-Deficient Human Cytomegalovirus by Epstein-Barr Virus Tegument Protein BNRF1.J Virol. 2016 Oct 14;90(21):9664-9673. doi: 10.1128/JVI.01224-16. Print 2016 Nov 1. J Virol. 2016. PMID: 27535048 Free PMC article.
-
Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection.J Virol. 2013 Feb;87(4):2174-85. doi: 10.1128/JVI.02950-12. Epub 2012 Dec 5. J Virol. 2013. PMID: 23221561 Free PMC article.
-
The replication defect of ICP0-null mutant herpes simplex virus 1 can be largely complemented by the combined activities of human cytomegalovirus proteins IE1 and pp71.J Virol. 2013 Jan;87(2):978-90. doi: 10.1128/JVI.01103-12. Epub 2012 Nov 7. J Virol. 2013. PMID: 23135716 Free PMC article.
-
Role of ND10 nuclear bodies in the chromatin repression of HSV-1.Virol J. 2016 Apr 5;13:62. doi: 10.1186/s12985-016-0516-4. Virol J. 2016. PMID: 27048561 Free PMC article. Review.
-
The use of fluorescence microscopy to study the association between herpesviruses and intrinsic resistance factors.Viruses. 2011 Dec;3(12):2412-24. doi: 10.3390/v3122412. Epub 2011 Dec 7. Viruses. 2011. PMID: 22355446 Free PMC article. Review.
Cited by
-
Interaction of herpes simplex virus ICP0 with ND10 bodies: a sequential process of adhesion, fusion, and retention.J Virol. 2013 Sep;87(18):10244-54. doi: 10.1128/JVI.01487-13. Epub 2013 Jul 17. J Virol. 2013. PMID: 23864622 Free PMC article.
-
Uncoupling uncoating of herpes simplex virus genomes from their nuclear import and gene expression.J Virol. 2011 May;85(9):4271-83. doi: 10.1128/JVI.02067-10. Epub 2011 Feb 23. J Virol. 2011. PMID: 21345968 Free PMC article.
-
SUMO Ligase Protein Inhibitor of Activated STAT1 (PIAS1) Is a Constituent Promyelocytic Leukemia Nuclear Body Protein That Contributes to the Intrinsic Antiviral Immune Response to Herpes Simplex Virus 1.J Virol. 2016 Jun 10;90(13):5939-5952. doi: 10.1128/JVI.00426-16. Print 2016 Jul 1. J Virol. 2016. PMID: 27099310 Free PMC article.
-
Chromatin dynamics during lytic infection with herpes simplex virus 1.Viruses. 2013 Jul 16;5(7):1758-86. doi: 10.3390/v5071758. Viruses. 2013. PMID: 23863878 Free PMC article. Review.
-
HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons.PLoS Pathog. 2012;8(8):e1002852. doi: 10.1371/journal.ppat.1002852. Epub 2012 Aug 9. PLoS Pathog. 2012. PMID: 22912575 Free PMC article.
References
-
- Amin, H. M., S. Saeed, and S. Alkan. 2001. Histone deacetylase inhibitors induce caspase-dependent apoptosis and downregulation of daxx in acute promyelocytic leukaemia with t(15;17). Br. J. Haematol. 115:287-297. - PubMed
-
- Argentaro, A., J. C. Yang, L. Chapman, M. S. Kowalczyk, R. J. Gibbons, D. R. Higgs, D. Neuhaus, and D. Rhodes. 2007. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc. Natl. Acad. Sci. U. S. A. 104:11939-11944. - PMC - PubMed
-
- Boddy, M. N., K. Howe, L. D. Etkin, E. Solomon, and P. S. Freemont. 1996. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 13:971-982. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
