

Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases
- PMID: 20147746
- PMCID: PMC2856980
- DOI: 10.1074/jbc.R109.072181
Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases
Abstract
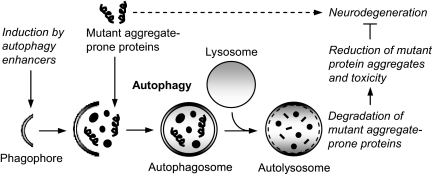
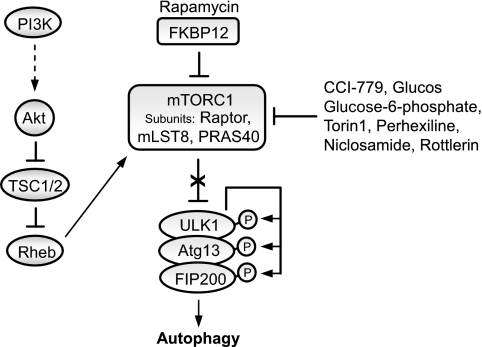
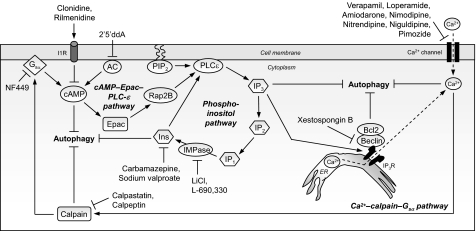
Many of the neurodegenerative diseases that afflict people are caused by intracytoplasmic aggregate-prone proteins. These include Parkinson disease, tauopathies, and polyglutamine expansion diseases such as Huntington disease. In Mendelian forms of these diseases, the mutations generally confer toxic novel functions on the relevant proteins. Thus, one potential strategy for dealing with these mutant proteins is to enhance their degradation. This can be achieved by up-regulating macroautophagy, which we will henceforth call autophagy. In this minireview, we will consider the reasons why autophagy up-regulation may be a powerful strategy for these diseases. In addition, we will consider some of the drugs and associated signaling pathways that can be used to induce autophagy with these therapeutic aims in mind.
Figures
References
-
- Rubinsztein D. C. (2006) Nature 443, 780–786 - PubMed
-
- Ross C. A., Poirier M. A. (2004) Nat. Med. 10, (suppl.) S10–S17 - PubMed
-
- Zoghbi H. Y., Orr H. T. (2000) Annu. Rev. Neurosci. 23, 217–247 - PubMed
-
- Imarisio S., Carmichael J., Korolchuk V., Chen C. W., Saiki S., Rose C., Krishna G., Davies J. E., Ttofi E., Underwood B. R., Rubinsztein D. C. (2008) Biochem. J. 412, 191–209 - PubMed
-
- Arrasate M., Mitra S., Schweitzer E. S., Segal M. R., Finkbeiner S. (2004) Nature 431, 805–810 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
