

Variable phenotypic expression of a MECP2 mutation in a family
- PMID: 20151026
- PMCID: PMC2819215
- DOI: 10.1007/s11689-009-9034-7
Variable phenotypic expression of a MECP2 mutation in a family
Abstract
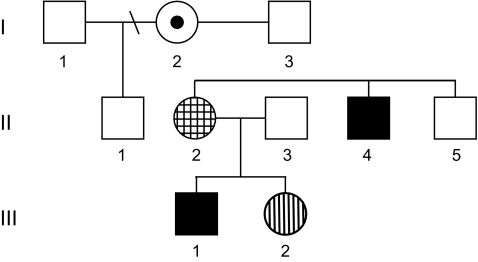
We report a three generation family in which five members, three females and two males, demonstrate a 44 bp deletion (1164-1207del44) in the MECP2 gene associated with Rett syndrome, leading to a truncation of the C-terminus of the protein. Two of the three females and both males do not meet RTT criteria whereas the youngest female has classic RTT. Both males demonstrated a clear pattern of progressive involvement including dystonia. The transmitting females do not demonstrate features of RTT as a result of unbalanced X chromosome inactivation (XCI) and were only identified as carriers following the evaluation of the affected males and the girl with classic RTT. As such, accurate assessment of the precise frequency of MECP2 mutations in carrier females with mild cognitive impairment or borderline cognitive function will be under-represented unless an affected offspring is recognized. Strategies for accurate diagnosis in such instances should be considered carefully.
Keywords: Dystonia; MECP2; Male; Mutation; Phenotype-genotype; Rett syndrome; X chromosome inactivation.
Figures
Similar articles
-
Two new Rett syndrome families and review of the literature: expanding the knowledge of MECP2 frameshift mutations.Orphanet J Rare Dis. 2011 Aug 30;6:58. doi: 10.1186/1750-1172-6-58. Orphanet J Rare Dis. 2011. PMID: 21878110 Free PMC article. Review.
-
Rett syndrome: the complex nature of a monogenic disease.J Mol Med (Berl). 2003 Jun;81(6):346-54. doi: 10.1007/s00109-003-0444-9. Epub 2003 May 16. J Mol Med (Berl). 2003. PMID: 12750821 Review.
-
[X chromosome inactivation patterns in patients with Rett syndrome and their mothers and the parental origin of the priority inactive X chromosome].Zhonghua Er Ke Za Zhi. 2006 Sep;44(9):648-52. Zhonghua Er Ke Za Zhi. 2006. PMID: 17217653 Chinese.
-
X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of rett syndrome.Am J Hum Genet. 2004 Mar;74(3):511-20. doi: 10.1086/382228. Epub 2004 Feb 17. Am J Hum Genet. 2004. PMID: 14973779 Free PMC article.
-
MECP2 mutations in Czech patients with Rett syndrome and Rett-like phenotypes: novel mutations, genotype-phenotype correlations and validation of high-resolution melting analysis for mutation scanning.J Hum Genet. 2016 Jul;61(7):617-25. doi: 10.1038/jhg.2016.19. Epub 2016 Mar 17. J Hum Genet. 2016. PMID: 26984561
Cited by
-
Progress in Rett Syndrome: from discovery to clinical trials.Wien Med Wochenschr. 2016 Sep;166(11-12):325-32. doi: 10.1007/s10354-016-0491-9. Epub 2016 Aug 4. Wien Med Wochenschr. 2016. PMID: 27491553 Free PMC article.
-
A perspective on "cure" for Rett syndrome.Orphanet J Rare Dis. 2018 Apr 2;13(1):44. doi: 10.1186/s13023-018-0786-6. Orphanet J Rare Dis. 2018. PMID: 29609636 Free PMC article.
-
Rett Syndrome: The Emerging Landscape of Treatment Strategies.CNS Drugs. 2024 Nov;38(11):851-867. doi: 10.1007/s40263-024-01106-y. Epub 2024 Sep 9. CNS Drugs. 2024. PMID: 39251501 Free PMC article. Review.
-
Rett Syndrome: Reaching for Clinical Trials.Neurotherapeutics. 2015 Jul;12(3):631-40. doi: 10.1007/s13311-015-0353-y. Neurotherapeutics. 2015. PMID: 25861995 Free PMC article. Review.
-
Two new Rett syndrome families and review of the literature: expanding the knowledge of MECP2 frameshift mutations.Orphanet J Rare Dis. 2011 Aug 30;6:58. doi: 10.1186/1750-1172-6-58. Orphanet J Rare Dis. 2011. PMID: 21878110 Free PMC article. Review.
References
-
- Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wiener Medizinische Wochenschrift. 1966;116:723–6. - PubMed
-
- Hagberg B, et al. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6(5):293–7. doi: 10.1053/ejpn.2002.0612. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials