

doi: 10.3390/v1030362.
The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors
Affiliations
- PMID: 20151027
- PMCID: PMC2819288
- DOI: 10.3390/v1030362
Item in Clipboard
The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors
Viruses.
2009.
Abstract
During the co-evolution of cytomegalovirus (CMV) and natural killer (NK) cells, each has evolved specific tactics in an attempt to prevail. CMV has evolved multiple immune evasion mechanisms to avoid detection by NK cells and other immune cells, leading to chronic infection. Meanwhile, the host has evolved virus-specific receptors to counter these evasion strategies. The natural selection of viral genes and host receptors allows us to observe a unique molecular example of "survival of the fittest", as virus and immune cells try to out-maneuver one another or for the virus to achieve détente for optimal dissemination in the population.
Figures
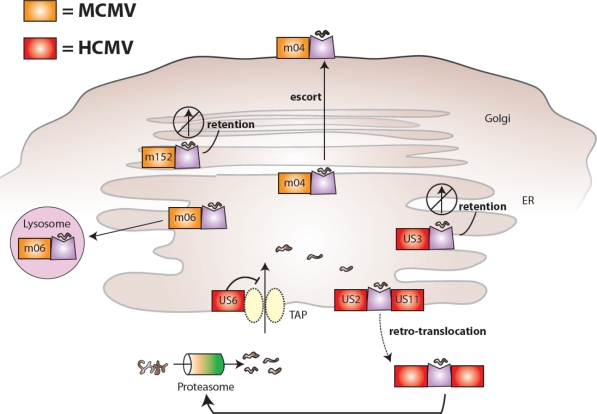
MCMV and HCMV proteins interfere with MHC class I presentation and host recognition of virally infected cells. MCMV-encoded glycoproteins (shown in orange) inhibit the biosynthesis and expression of mouse MHC class I molecules: m06 binds MHC class I in the endoplasmic reticulum (ER) and redirects it to lysosomes leading to degradation, m152 retains MHC class I in an ER-to-Golgi intermediate compartment, and m04 forms a complex with MHC class I in the ER and together they are transported to the cell surface. HCMV-encoded glycoproteins (shown in red) also inhibit the biosynthesis and expression of human MHC class I molecules: US6 prevents translocation of proteasome-generated peptides from entering the ER via TAP (a specific transporter of peptides for loading on MHC class I molecules, associated with antigen presentation), US3 retains MHC class I in the ER, and US2 and US11 both bind MHC class I in the ER and mediate retro-translocation of molecules back to the cytoplasm via the Sec61 channel leading to proteasomal degradation.
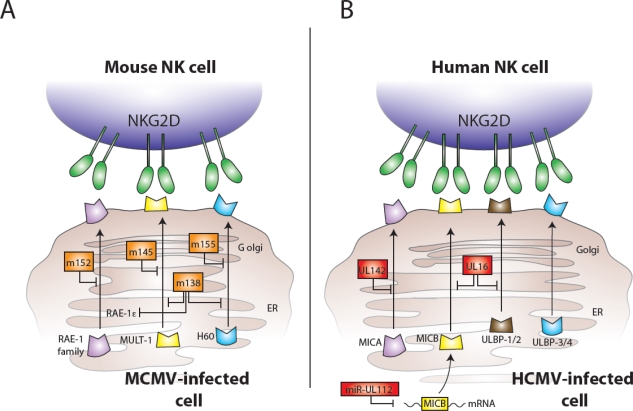
MCMV and HCMV proteins interfere with expression of NKG2D ligands and host recognition of virally infected cells. (A) MCMV-encoded glycoproteins (shown in orange) inhibit the expression of mouse NKG2D ligands: m152 interferes with expression of all five members of the RAE-1 family, m145 prevents surface expression of MULT1, m155 causes degradation of H60, and m138 assists to block expression of RAE-1ε, MULT1, and H60. (B) HCMV-encoded components (shown in red) also inhibit the expression of NKG2D ligands: UL142 inhibits MICA expression, UL16 binds MICB, ULBP1, and ULBP2 in the Golgi, and the miR-UL112 microRNA targets MICB mRNA for degradation leading to diminished cell surface expression of MICB.
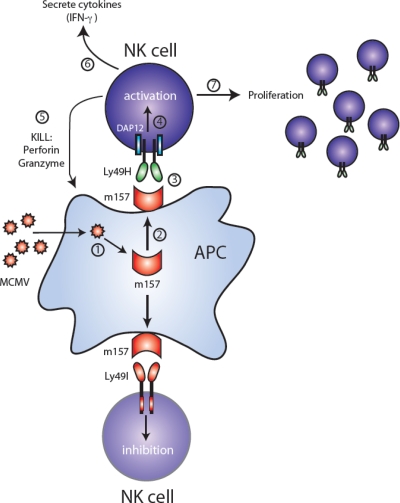
MCMV-encoded m157 leads to activation or inhibition depending on NK cell receptor expression. Following MCMV infection of a target cell (step 1), the m157 glycoprotein is expressed (step 2). NK cells from C57BL/6 mice express the activating Ly49H receptor (top half of figure), which associates with and signals through the ITAM-containing DAP12 adaptor molecule. Recognition of m157 by Ly49H-bearing NK cells (step 3) leads to activation via DAP12 (step 4), followed by cell-mediated cytotoxicity via perforin and granzymes (step 5), secretion of cytokines such as IFN-γ (step 6), and a clonal-like proliferation (step 7). In contrast, NK cells from 129/J mice express the ITIM-containing Ly49I receptor (bottom half of figure), which binds m157 on MCMV-infected cells, and become inhibited.
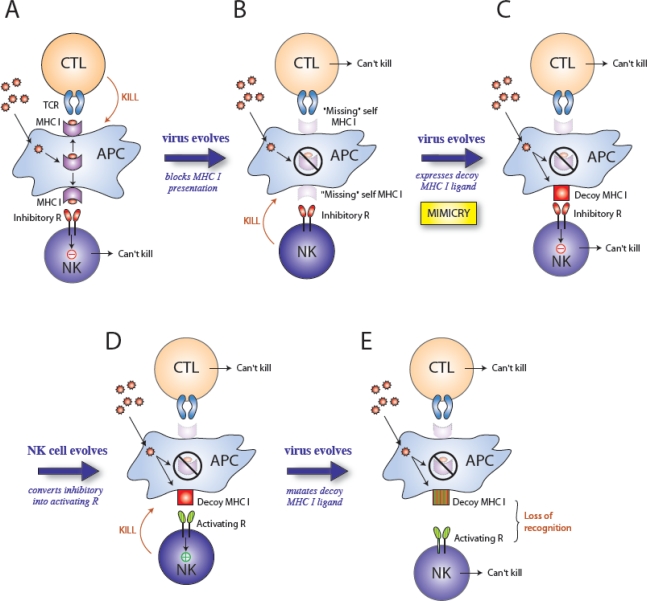
Selective pressures drive the evolution of virus and host genes. A model predicting the influence of natural selection on evolution of herpesviruses and NK cell receptors: (A) During infection, viral peptides presented on MHC class I are recognized by CD8+ T cells (CTL) and the infected cell is killed. (B) The virus evades the host immune response by evolving mechanisms to block MHC class I presentation. Although the CTL response is ineffective due to loss of antigen presentation, NK cells sense “missing self” (lack of MHC class I) and kill the infected cell because they are not inhibited. (C) The virus evades NK cell surveillance by the acquisition of a MHC class I decoy molecules (i.e. molecular mimicry), which bind inhibitory receptors on NK cells. Neither the CTL nor NK cell can kill the infected cell. (D) The host immune system evolves. NK cells acquire an activating receptor that recognizes the viral MHC class I decoy via duplication and gene conversion of the inhibitory receptor; NK cells now specifically recognize the viral component and kill the infected cell. (E) The virus evolves and continues to evade the host immune system, by mutating the decoy MHC class I molecule so that NK cells can no longer recognize or kill the infected cell.
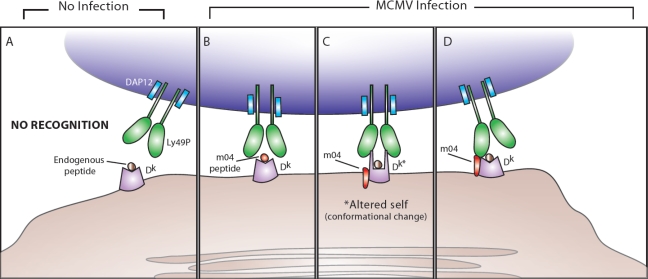
Possible mechanisms behind Ly49P recognition of m04 and H2-Dk during MCMV infection. (A) In the absence of MCMV infection, DAP12-associated Ly49P does not interact with MHC class I H2-Dk presenting endogenous peptides. (B) During MCMV infection, Ly49P-bearing NK cells might recognize a m04-derived peptide associated with H2-Dk. (C) During MCMV infection, Ly49P might interact with a H2-Dk molecule that has undergone a conformational change because of m04 binding (the altered self-MHC class I is labeled in the diagram as Dk*). (D) During MCMV infection, Ly49P-bearing NK cells might recognize the entire m04/H2-Dk complex, with the extracellular domains of Ly49P interacting with portions of both m04 and H2-Dk.
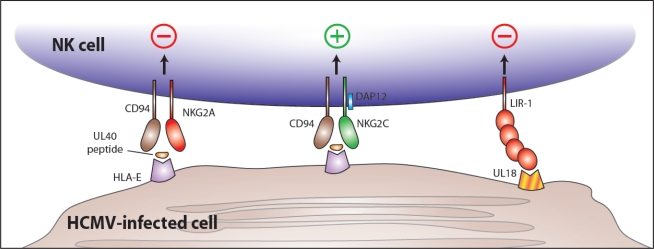
NK cell recognition of HCMV-encoded proteins during infection. The HCMV-encoded UL40 protein contains an epitope (from the UL40 leader sequence) that is bound by HLA-E and presented at the surface of virally infected cells. Binding of UL40 peptide/HLA-E by the inhibitory CD94/NKG2A receptor leading to NK cell inhibition could represent an immune evasion mechanism of HCMV. However, the activating CD94/NKG2C receptor might also engage the UL40 peptide/HLA-E complex (although with lower affinity) leading to NK cell activation. At the same time, the HCMV-encoded UL18 glycoprotein is expressed at the surface of infected cells and binds LIR-1, an inhibitory receptor, leading to inhibition of NK cell responses. (There is also evidence suggesting that the CD94/NKG2C may also recognize UL18, although a functional consequence for such an interaction is currently lacking.)
Similar articles
-
Hematopoietic cell-mediated dissemination of murine cytomegalovirus is regulated by NK cells and immune evasion.PLoS Pathog. 2021 Jan 28;17(1):e1009255. doi: 10.1371/journal.ppat.1009255. eCollection 2021 Jan. PLoS Pathog. 2021. PMID: 33508041 Free PMC article.
-
All is fair in virus-host interactions: NK cells and cytomegalovirus.Trends Mol Med. 2011 Nov;17(11):677-85. doi: 10.1016/j.molmed.2011.07.003. Epub 2011 Aug 17. Trends Mol Med. 2011. PMID: 21852192 Free PMC article.
-
How the virus outsmarts the host: function and structure of cytomegalovirus MHC-I-like molecules in the evasion of natural killer cell surveillance.J Biomed Biotechnol. 2011;2011:724607. doi: 10.1155/2011/724607. Epub 2011 Jun 30. J Biomed Biotechnol. 2011. PMID: 21765638 Free PMC article. Review.
-
Deciphering the Immunological Phenomenon of Adaptive Natural Killer (NK) Cells and Cytomegalovirus (CMV).Int J Mol Sci. 2020 Nov 23;21(22):8864. doi: 10.3390/ijms21228864. Int J Mol Sci. 2020. PMID: 33238550 Free PMC article. Review.
-
DNAM-1 Activating Receptor and Its Ligands: How Do Viruses Affect the NK Cell-Mediated Immune Surveillance during the Various Phases of Infection?Int J Mol Sci. 2019 Jul 30;20(15):3715. doi: 10.3390/ijms20153715. Int J Mol Sci. 2019. PMID: 31366013 Free PMC article. Review.
Cited by
-
Natural Killer Cell Evasion Is Essential for Infection by Rhesus Cytomegalovirus.PLoS Pathog. 2016 Aug 31;12(8):e1005868. doi: 10.1371/journal.ppat.1005868. eCollection 2016 Aug. PLoS Pathog. 2016. PMID: 27580123 Free PMC article.
-
Immune memory redefined: characterizing the longevity of natural killer cells.Immunol Rev. 2010 Jul;236:83-94. doi: 10.1111/j.1600-065X.2010.00900.x. Immunol Rev. 2010. PMID: 20636810 Free PMC article. Review.
-
Virus encoded MHC-like decoys diversify the inhibitory KIR repertoire.PLoS Comput Biol. 2013;9(10):e1003264. doi: 10.1371/journal.pcbi.1003264. Epub 2013 Oct 10. PLoS Comput Biol. 2013. PMID: 24130473 Free PMC article.
-
Killer Immunoglobulin-like Receptors (KIR) haplogroups A and B track with Natural Killer Cells and Cytokine Profile in Aged Subjects: Observations from Octo/Nonagenarians in the Belfast Elderly Longitudinal Free-living Aging STudy (BELFAST).Immun Ageing. 2013 Aug 19;10(1):35. doi: 10.1186/1742-4933-10-35. Immun Ageing. 2013. PMID: 23957956 Free PMC article.
-
CD1d Expression and Invariant NKT Cell Responses in Herpesvirus Infections.Front Immunol. 2015 Jun 25;6:312. doi: 10.3389/fimmu.2015.00312. eCollection 2015. Front Immunol. 2015. PMID: 26161082 Free PMC article. Review.
References
-
- Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. - PubMed
-
- Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. - PubMed
-
- Gilbert SC, Plebanski M, Gupta S, Morris J, Cox M, Aidoo M, Kwiatkowski D, Greenwood BM, Whittle HC, Hill AV. Association of malaria parasite population structure, HLA, and immunological antagonism. Science. 1998;279:1173–1177. - PubMed
-
- Hill AV, Allsopp CE, Kwiatkowski D, Anstey NM, Twumasi P, Rowe PA, Bennett S, Brewster D, McMichael AJ, Greenwood BM. Common west African HLA antigens are associated with protection from severe malaria. Nature. 1991;352:595–600. - PubMed
-
- Mocarski ES., Jr Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 2002;10:332–339. - PubMed
