

Sindbis viral vector induced apoptosis requires translational inhibition and signaling through Mcl-1 and Bak
- PMID: 20152035
- PMCID: PMC2843653
- DOI: 10.1186/1476-4598-9-37
Sindbis viral vector induced apoptosis requires translational inhibition and signaling through Mcl-1 and Bak
Abstract
Background: Sindbis viral vectors are able to efficiently target and kill tumor cells in vivo, as shown using pancreatic and ovarian cancer models. Infection results in apoptosis both in vitro and in vivo. Sindbis vector uptake is mediated by the LAMR, which is upregulated on a number of different tumor types, thus conferring specificity of the vector to a wide range of cancers. In this study we elucidate the mechanism of apoptosis in two tumor cell lines, MOSEC, derived from the ovarian epithelium and Pan02, derived from a pancreatic adenocarcinoma. A comprehensive understanding of the mechanism of apoptosis would facilitate the design of more effective vectors for cancer therapy.
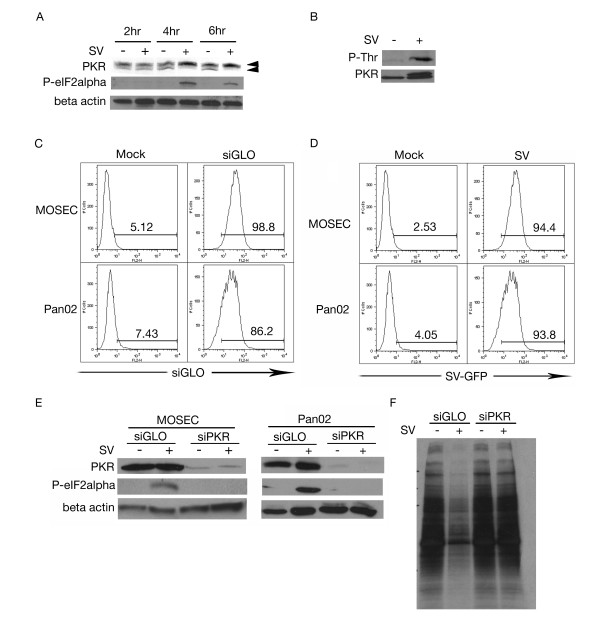
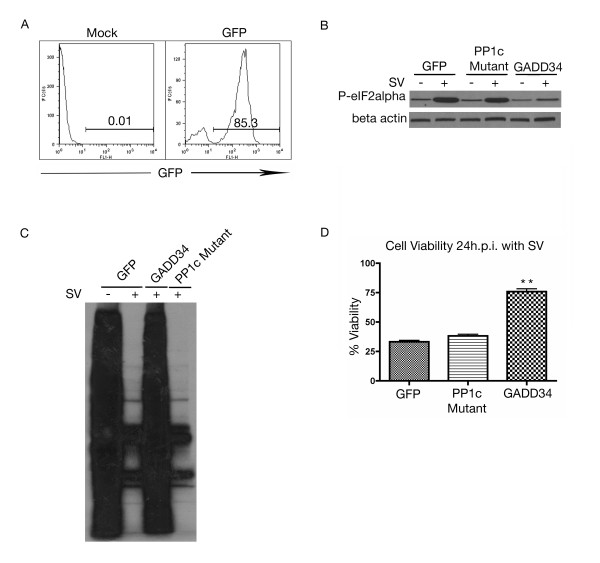
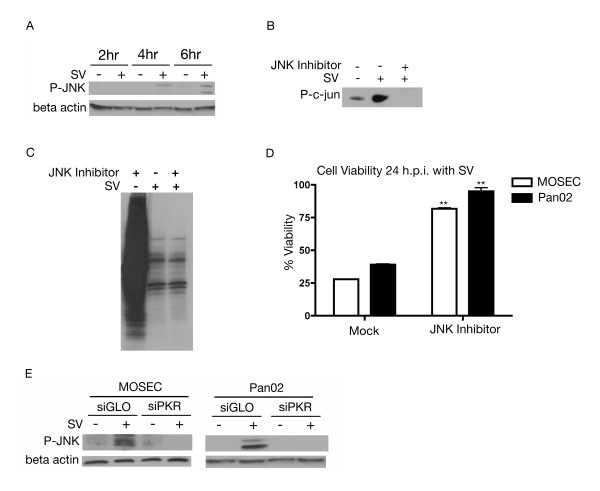
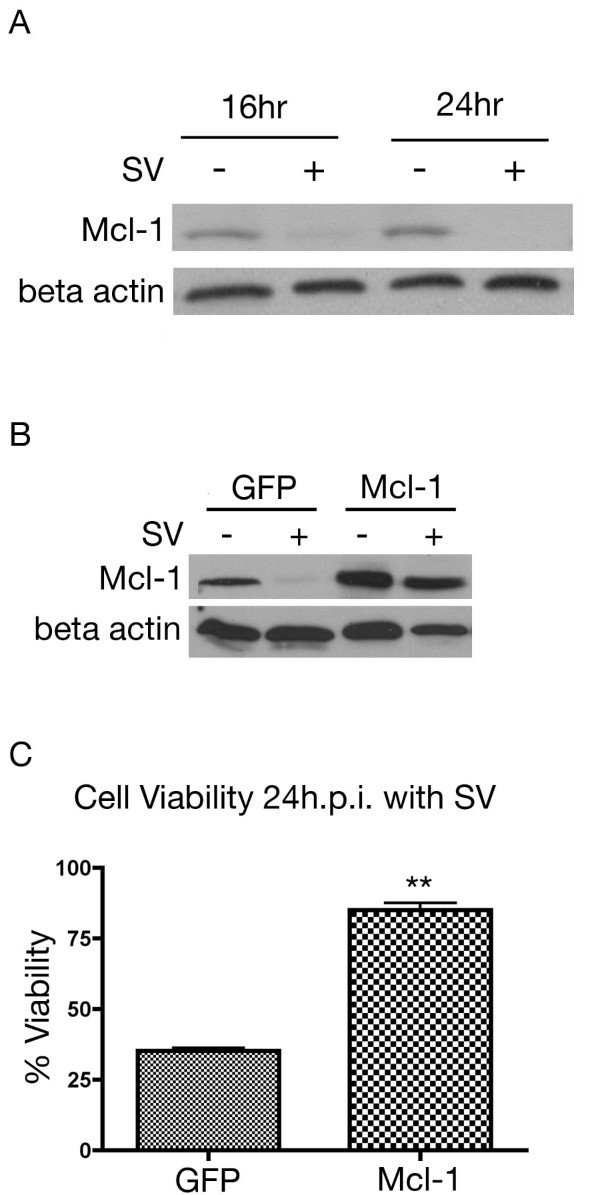
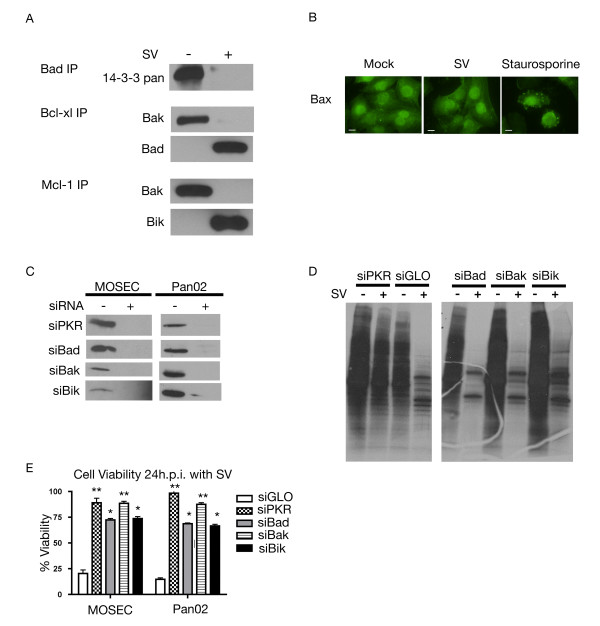
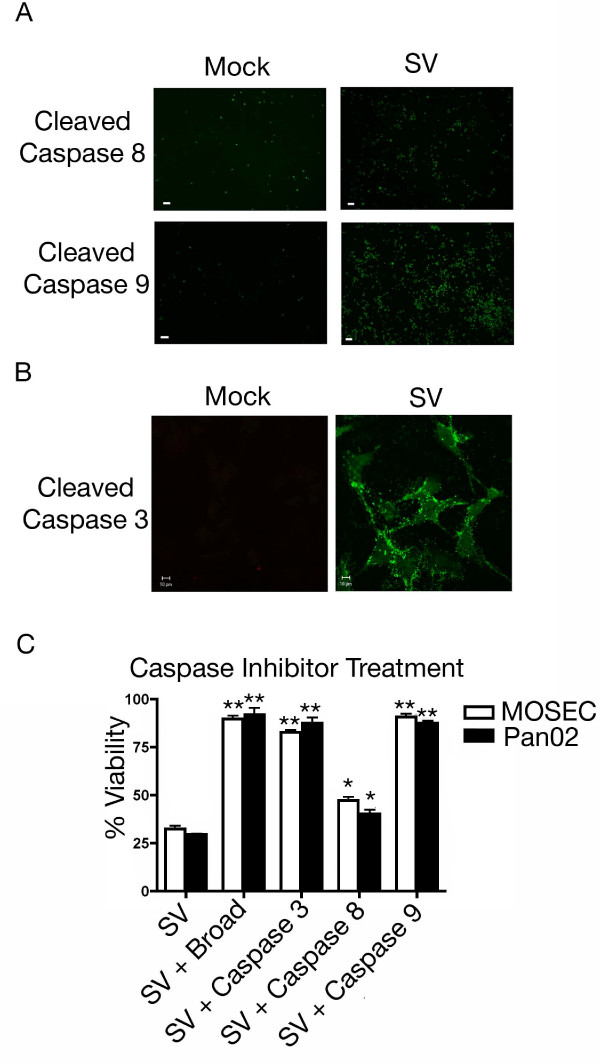
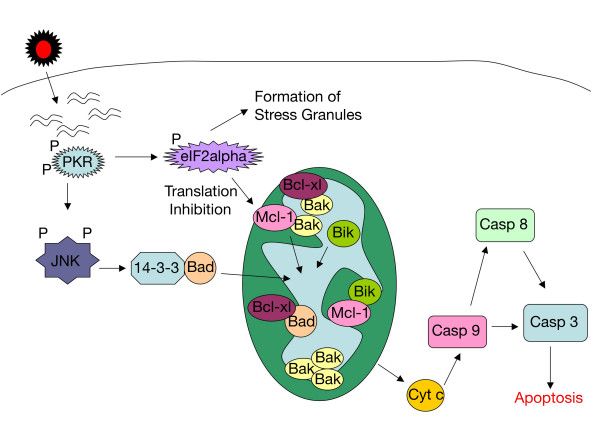
Results: The initial phase of Sindbis vector induced apoptosis in MOSEC and Pan02 models reconfirms that viral infection is sensed by PKR due to double-stranded RNA intermediates associated with genomic replication. PKR activation results in translation inhibition through eIF2alpha phosphorylation and initiation of the stress response. Our studies indicate that the roles of two proteins, Mcl-1 and JNK, intimately link Sindbis induced translational arrest and cellular stress. Translational arrest inhibits the synthesis of anti-apoptotic Bcl-2 protein, Mcl-1. JNK activation triggers the release of Bad from 14-3-3, which ultimately results in apoptosis. These signals from translational arrest and cellular stress are propagated to the mitochondria where Bad and Bik bind to Bcl-xl and Mcl-1 respectively. Formation of these heterodimers displaces Bak, which results in caspase 9 cleavage and signaling through the mitochondrial pathway of apoptosis.
Conclusion: The host cell response to Sindbis is triggered through PKR activation. Our studies demonstrate that PKR activation and subsequent translational arrest is linked to both cellular stress and apoptosis. We have also found the linkage point between translational arrest and apoptosis to be Mcl-1, a protein whose constant translation is required for inhibition of apoptosis. With this information vectors can be designed, which express or repress proteins implicated in this study, to enhance their therapeutic potential.
Figures

Similar articles
-
A novel BH3 mimetic efficiently induces apoptosis in melanoma cells through direct binding to anti-apoptotic Bcl-2 family proteins, including phosphorylated Mcl-1.Pigment Cell Melanoma Res. 2015 Mar;28(2):161-70. doi: 10.1111/pcmr.12325. Epub 2014 Dec 18. Pigment Cell Melanoma Res. 2015. PMID: 25324174
-
NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition.Genes Dev. 2007 Apr 15;21(8):929-41. doi: 10.1101/gad.1522007. Epub 2007 Apr 2. Genes Dev. 2007. PMID: 17403773 Free PMC article.
-
NF-κB Activation Promotes Alphavirus Replication in Mature Neurons.J Virol. 2019 Nov 26;93(24):e01071-19. doi: 10.1128/JVI.01071-19. Print 2019 Dec 15. J Virol. 2019. PMID: 31554691 Free PMC article.
-
Neuronal apoptosis pathways in Sindbis virus encephalitis.Prog Mol Subcell Biol. 2004;36:71-93. doi: 10.1007/978-3-540-74264-7_5. Prog Mol Subcell Biol. 2004. PMID: 15171608 Review.
-
Mcl-1 Protein and Viral Infections: A Narrative Review.Int J Mol Sci. 2024 Jan 17;25(2):1138. doi: 10.3390/ijms25021138. Int J Mol Sci. 2024. PMID: 38256213 Free PMC article. Review.
Cited by
-
The Regulation of Integrated Stress Response Signaling Pathway on Viral Infection and Viral Antagonism.Front Microbiol. 2022 Feb 11;12:814635. doi: 10.3389/fmicb.2021.814635. eCollection 2021. Front Microbiol. 2022. PMID: 35222313 Free PMC article. Review.
-
Potent and Targeted Sindbis Virus Platform for Immunotherapy of Ovarian Cancer.Cells. 2022 Dec 24;12(1):77. doi: 10.3390/cells12010077. Cells. 2022. PMID: 36611875 Free PMC article.
-
Channeling the Natural Properties of Sindbis Alphavirus for Targeted Tumor Therapy.Int J Mol Sci. 2023 Oct 6;24(19):14948. doi: 10.3390/ijms241914948. Int J Mol Sci. 2023. PMID: 37834397 Free PMC article. Review.
-
Apoptosis, autophagy and unfolded protein response pathways in Arbovirus replication and pathogenesis.Expert Rev Mol Med. 2016 Jan 19;18:e1. doi: 10.1017/erm.2015.19. Expert Rev Mol Med. 2016. PMID: 26781343 Free PMC article. Review.
-
Replicase-based plasmid DNA shows anti-tumor activity.BMC Cancer. 2011 Mar 28;11:110. doi: 10.1186/1471-2407-11-110. BMC Cancer. 2011. PMID: 21443770 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
