

Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy
- PMID: 20152563
- PMCID: PMC2852685
- DOI: 10.1016/j.jacc.2009.11.020
Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy
Erratum in
- J Am Coll Cardiol. 2010 Mar 30;55(13):1401. Pillichou, Kalliopi [corrected to Pilichou, Kalliopi]
Abstract
Objectives: The aim of this study was to define the genetic basis of arrhythmogenic right ventricular cardiomyopathy (ARVC).
Background: Arrhythmogenic right ventricular cardiomyopathy, characterized by right ventricular fibrofatty replacement and arrhythmias, causes sudden death. Autosomal dominant inheritance, reduced penetrance, and 7 desmosome-encoding causative genes are known. The basis of low penetrance is poorly understood.
Methods: Arrhythmogenic right ventricular cardiomyopathy probands and family members were enrolled, blood was obtained, lymphoblastoid cell lines were immortalized, deoxyribonucleic acid was extracted, polymerase chain reaction (PCR) amplification of desmosome-encoding genes was performed, PCR products were sequenced, and diseased tissue samples were studied for intercellular junction protein distribution with confocal immunofluorescence microscopy and antibodies against key proteins.
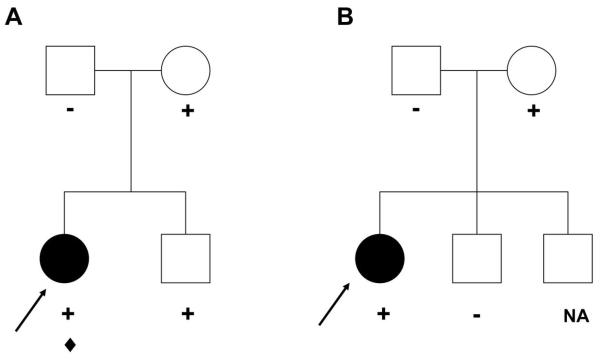
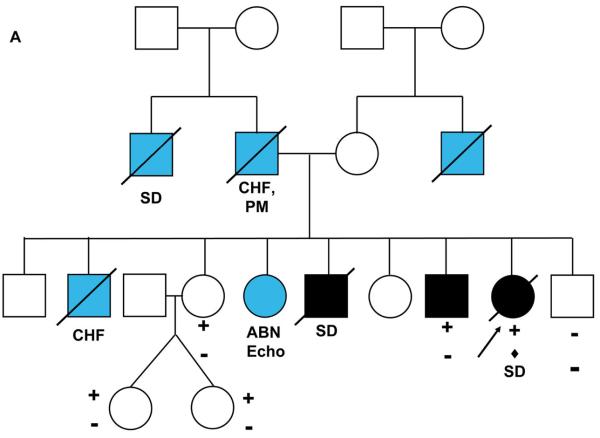
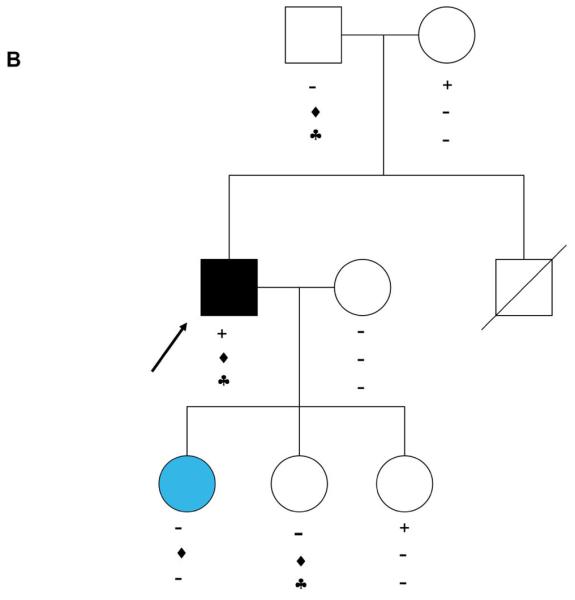
Results: We identified 21 variants in plakophilin-2 (PKP2) in 38 of 198 probands (19%), including missense, nonsense, splice site, and deletion/insertion mutations. Pedigrees showed wide intra-familial variability (severe early-onset disease to asymptomatic individuals). In 9 of 38 probands, PKP2 variants were identified that were encoded in trans (compound heterozygosity). The 38 probands hosting PKP2 variants were screened for other desmosomal genes mutations; second variants (digenic heterozygosity) were identified in 16 of 38 subjects with PKP2 variants (42%), including desmoplakin (DSP) (n = 6), desmoglein-2 (DSG2) (n = 5), plakophilin-4 (PKP4) (n = 1), and desmocollin-2 (DSC2) (n = 1). Heterozygous mutations in non-PKP 2 desmosomal genes occurred in 14 of 198 subjects (7%), including DSP (n = 4), DSG2 (n = 5), DSC2 (n = 3), and junctional plakoglobin (JUP) (n = 2). All variants occurred in conserved regions; none was identified in 700 ethnic-matched control subjects. Immunohistochemical analysis demonstrated abnormalities of protein architecture.
Conclusions: These data suggest that the genetic basis of ARVC includes reduced penetrance with compound and digenic heterozygosity. Disturbed junctional cytoarchitecture in subjects with desmosomal mutations confirms that ARVC is a disease of the desmosome and cell junction.
Copyright 2010 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–398. - PubMed
-
- Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J. Am. Coll. Cardiol. 1997;30:1512–1520. - PubMed
-
- Horimoto M, Funayama N, Satoh M, Igarashi T, Sekiguchi M. Histologic evidence of left ventricular involvement in arrhythmogenic right ventricular dysplasia. Jpn Circ J. 1989;53:1530–1534. - PubMed
-
- Towbin JA. Molecular genetics of sudden cardiac death. Cardiovasc Pathol. 2001;10:283–295. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
