

Distinct role of Kruppel-like factor 11 in the regulation of prostaglandin E2 biosynthesis
- PMID: 20154088
- PMCID: PMC2857022
- DOI: 10.1074/jbc.M109.077065
Distinct role of Kruppel-like factor 11 in the regulation of prostaglandin E2 biosynthesis
Abstract
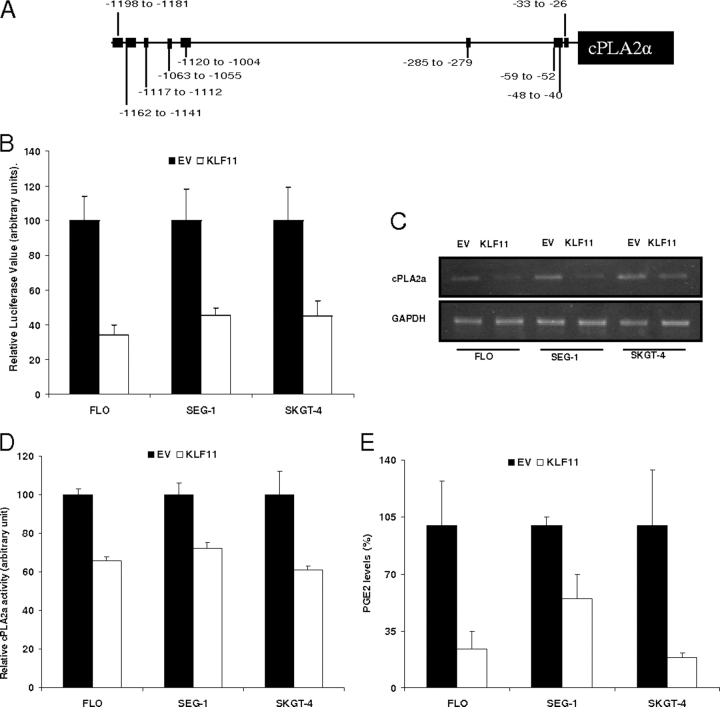
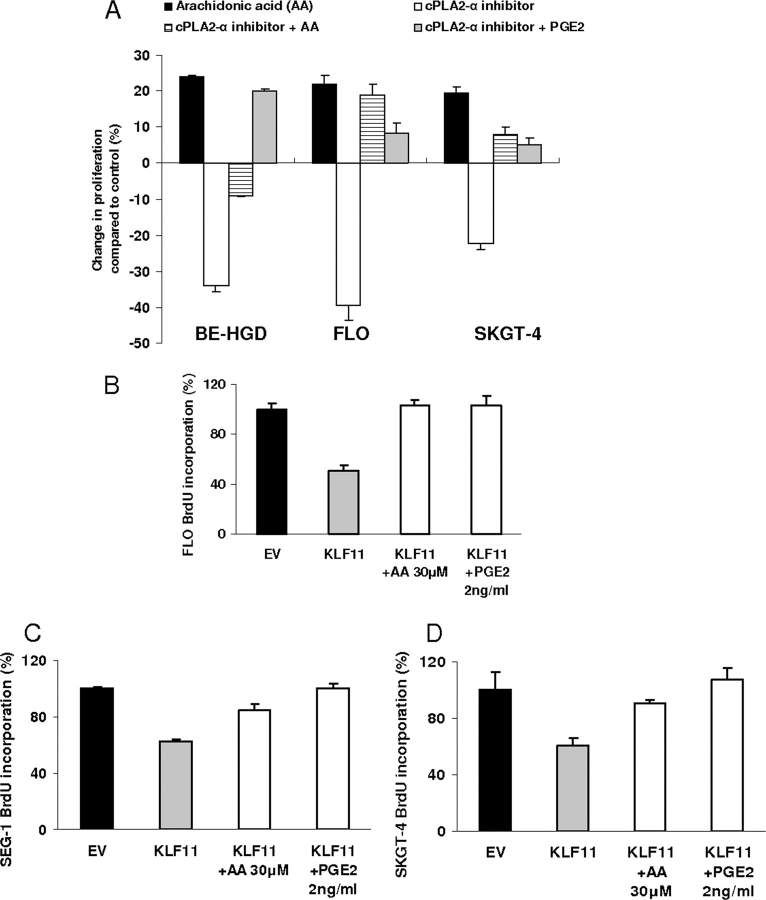
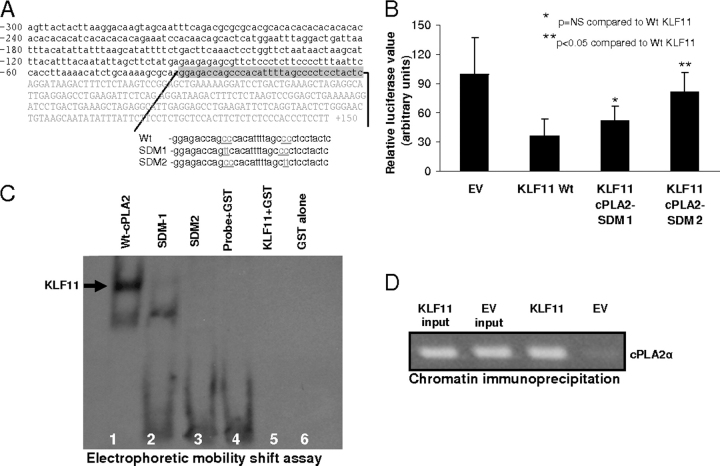
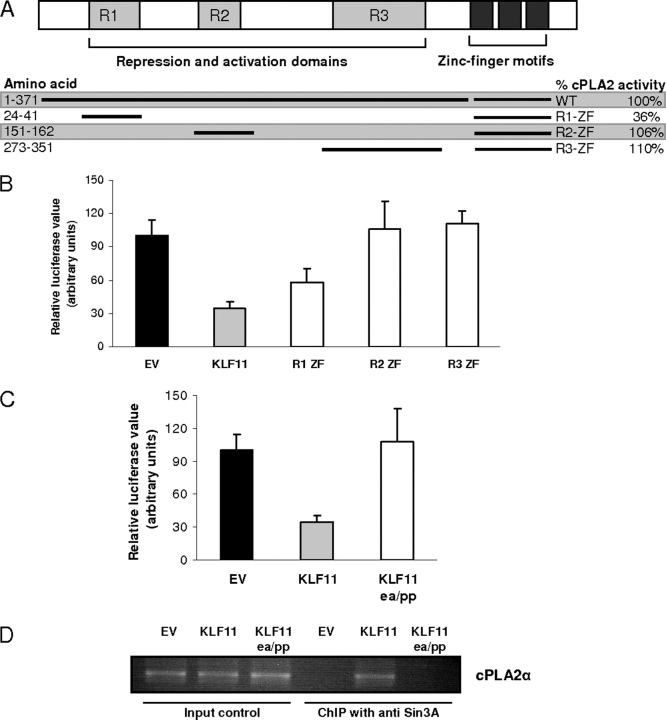
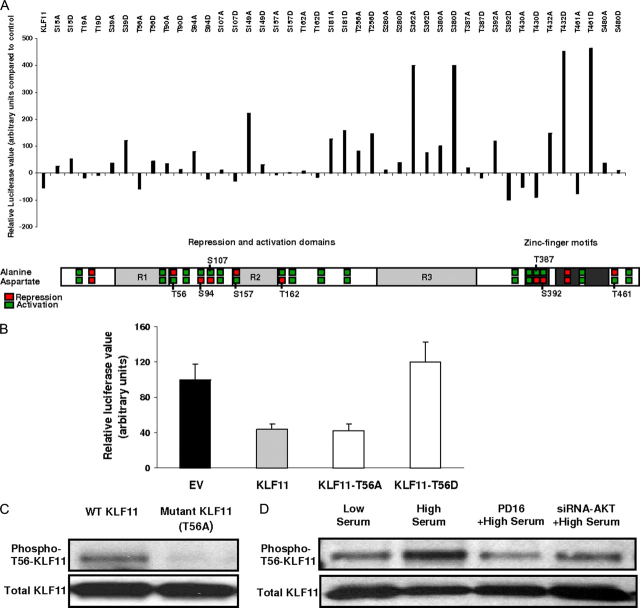
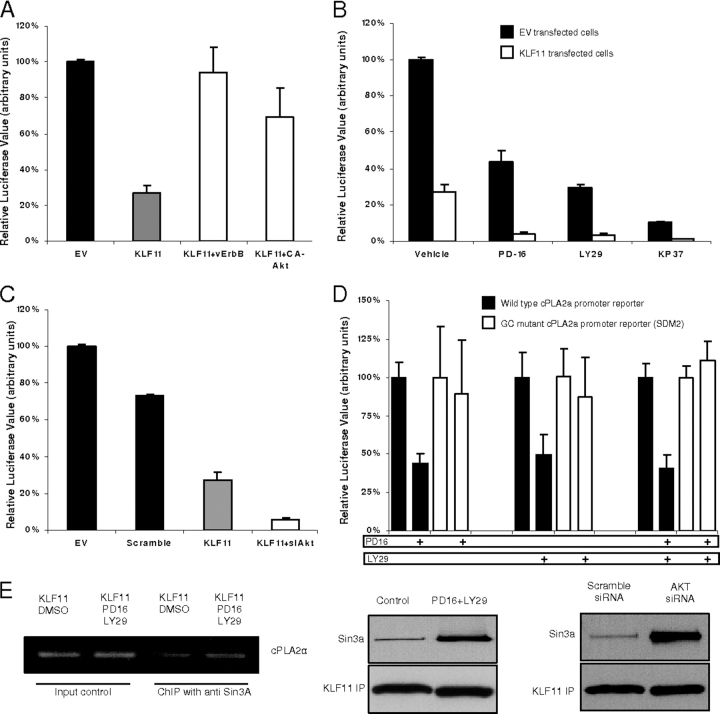
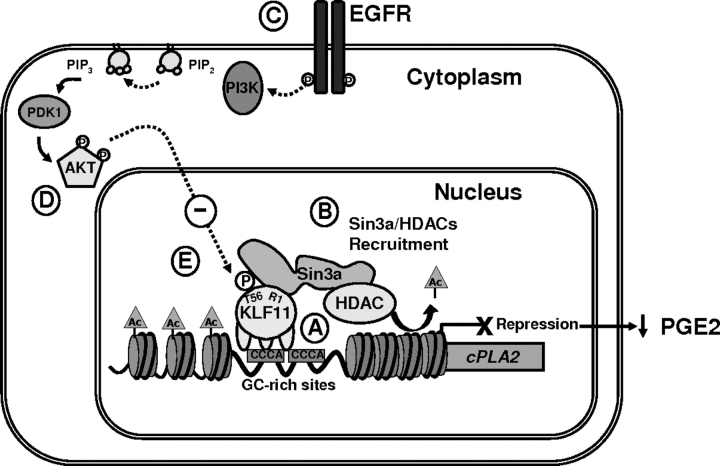
Kruppel-like factor (KLF) proteins are emerging as key regulators of lipid metabolism, diabetes, and the biosynthesis of immunological cytokines. However, their role in the synthesis of prostaglandins, widely known biochemical mediators that act in a myriad of cell biological processes remain poorly understood. Consequently, in this study a comprehensive investigation at the cellular, biochemical, and molecular levels reveal that KLF11 inhibits prostaglandin E(2) synthesis via transcriptional silencing of the promoter of its biosynthetic enzyme, cytosolic phospholipase A2alpha. Mechanistically, KLF11 accomplishes this function by binding to the promoter via specific GC-rich sites and recruiting the Sin3-histone deacetylase chromatin remodeling complex. Further functional characterization reveals that this function of KLF11 can be reversed by epidermal growth factor receptor-AKT-mediated post-translational modification of threonine 56, a residue within its Sin3-binding domain. This is the first evidence supporting a relevant role for any KLF protein in doing both: transcriptionally inhibiting prostaglandin biosynthesis and its reversibility by an epidermal growth factor receptor-AKT signaling-mediated posttranslational mechanisms.
Figures
References
-
- Murakami M., Kudo I. Prog. Lipid Res. 2004;43:3–35. - PubMed
-
- Murakami M., Shimbara S., Kambe T., Kuwata H., Winstead M.V., Tischfield J.A., Kudo I. J. Biol. Chem. 1998;273:14411–14423. - PubMed
-
- Leslie C.C. J. Biol. Chem. 1997;272:16709–16712. - PubMed
-
- Funk C.D. Science. 2001;294:1871–1875. - PubMed
-
- Bonventre J.V., Huang Z., Taheri M.R., O'Leary E., Li E., Moskowitz M.A., Sapirstein A. Nature. 1997;390:622–625. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
