

BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells
- PMID: 20154269
- PMCID: PMC2874480
- DOI: 10.1096/fj.09-138305
BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells
Abstract
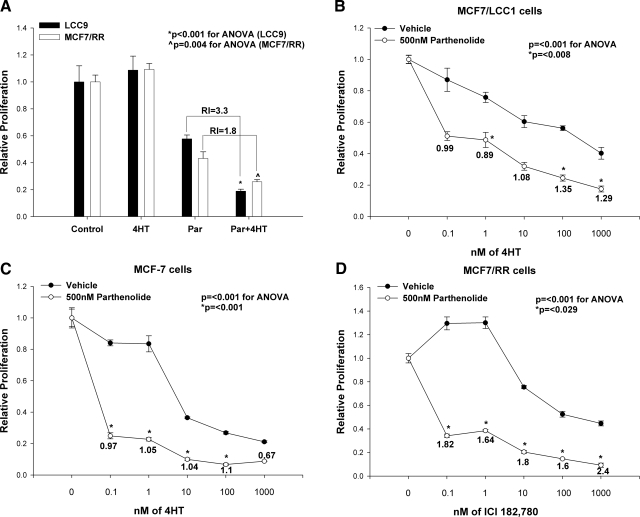
Resistance to endocrine therapies remains a major problem in the management of estrogen receptor-alpha (ER)-positive breast cancer. We show that inhibition of NF-kappaB (p65/RELA), either by overexpression of a mutant IkappaB (IkappaBSR) or a small-molecule inhibitor of NF-kappaB (parthenolide; IC(50)=500 nM in tamoxifen-resistant cells), synergistically restores sensitivity to 4-hydroxytamoxifen (4HT) in resistant MCF7/RR and MCF7/LCC9 cells and further sensitizes MCF-7 and MCF7/LCC1 control cells to 4HT. These effects are independent of changes in either cell cycle distribution or in the level of autophagy measured by inhibition of p62/SQSTM1 expression and cleavage of LC3. NF-kappaB inhibition restores the ability of 4HT to decrease BCL2 expression, increase mitochondrial membrane permeability, and induce a caspase-dependent apoptotic cell death in resistant cells. Each of these effects is reversed by a caspase 8 (CASP8)-specific inhibitor that blocks enzyme-substrate binding. Thus, increased activation of NF-kappaB can alter sensitivity to tamoxifen by modulating CASP8 activity, with consequent effects on BCL2 expression, mitochondrial function, and apoptosis. These data provide significant new insights into how molecular signaling affects antiestrogen responsiveness and strongly suggest that a combination of parthenolide and tamoxifen may offer a novel therapeutic approach to the management of some ER-positive breast cancers.
Figures










Similar articles
-
IFNgamma restores breast cancer sensitivity to fulvestrant by regulating STAT1, IFN regulatory factor 1, NF-kappaB, BCL2 family members, and signaling to caspase-dependent apoptosis.Mol Cancer Ther. 2010 May;9(5):1274-85. doi: 10.1158/1535-7163.MCT-09-1169. Mol Cancer Ther. 2010. PMID: 20457620 Free PMC article.
-
Garcinol, an acetyltransferase inhibitor, suppresses proliferation of breast cancer cell line MCF-7 promoted by 17β-estradiol.Asian Pac J Cancer Prev. 2014;15(12):5001-7. doi: 10.7314/apjcp.2014.15.12.5001. Asian Pac J Cancer Prev. 2014. PMID: 24998578
-
Anterior gradient-2 plays a critical role in breast cancer cell growth and survival by modulating cyclin D1, estrogen receptor-alpha and survivin.Breast Cancer Res. 2010;12(3):R32. doi: 10.1186/bcr2586. Epub 2010 Jun 4. Breast Cancer Res. 2010. PMID: 20525379 Free PMC article.
-
NF-κB signaling in therapy resistance of breast cancer: Mechanisms, approaches, and challenges.Life Sci. 2024 Jul 1;348:122684. doi: 10.1016/j.lfs.2024.122684. Epub 2024 May 4. Life Sci. 2024. PMID: 38710275 Review.
-
p62 at the crossroads of autophagy, apoptosis, and cancer.Cell. 2009 Jun 12;137(6):1001-4. doi: 10.1016/j.cell.2009.05.023. Cell. 2009. PMID: 19524504 Free PMC article. Review.
Cited by
-
Down-regulation of Forkhead box protein A1 (FOXA1) leads to cancer stem cell-like properties in tamoxifen-resistant breast cancer cells through induction of interleukin-6.J Biol Chem. 2017 May 19;292(20):8136-8148. doi: 10.1074/jbc.M116.763276. Epub 2017 Mar 7. J Biol Chem. 2017. PMID: 28270510 Free PMC article.
-
Genomics of the NF-κB signaling pathway: hypothesized role in ovarian cancer.Cancer Causes Control. 2011 May;22(5):785-801. doi: 10.1007/s10552-011-9745-4. Epub 2011 Feb 27. Cancer Causes Control. 2011. PMID: 21359843 Free PMC article. Review.
-
When is a vesicle not just a vesicle: mitochondrial spheroids and mitochondrial autophagosomes.Cell Biosci. 2014 Nov 14;4:66. doi: 10.1186/2045-3701-4-66. eCollection 2014. Cell Biosci. 2014. PMID: 25699171 Free PMC article. No abstract available.
-
COUP-TFII inhibits NFkappaB activation in endocrine-resistant breast cancer cells.Mol Cell Endocrinol. 2014 Jan 25;382(1):358-367. doi: 10.1016/j.mce.2013.10.010. Epub 2013 Oct 17. Mol Cell Endocrinol. 2014. PMID: 24141032 Free PMC article.
-
The different roles of ER subtypes in cancer biology and therapy.Nat Rev Cancer. 2011 Jul 22;11(8):597-608. doi: 10.1038/nrc3093. Nat Rev Cancer. 2011. PMID: 21779010 Review.
References
-
- Clarke R, Leonessa F, Welch J N, Skaar T C. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53:25–71. - PubMed
-
- Clarke R, Liu M C, Bouker K B, Gu Z, Lee R Y, Zhu Y, Skaar T C, Gomez B, O'Brien K, Wang Y, Hilakivi-Clarke L A. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22:7316–7339. - PubMed
-
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun M J. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. - PubMed
-
- Riggins R, Bouton A H, Liu M C, Clarke R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitam Horm. 2005;71:201–237. - PubMed
-
- Encarnacion C A, Ciocca D R, McGuire W L, Clark G M, Fuqua S A, Osborne C K. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat. 1993;26:237–246. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
