

Inflammation and insulin resistance induced by trans-10, cis-12 conjugated linoleic acid depend on intracellular calcium levels in primary cultures of human adipocytes
- PMID: 20154361
- PMCID: PMC2882722
- DOI: 10.1194/jlr.M005447
Inflammation and insulin resistance induced by trans-10, cis-12 conjugated linoleic acid depend on intracellular calcium levels in primary cultures of human adipocytes
Abstract
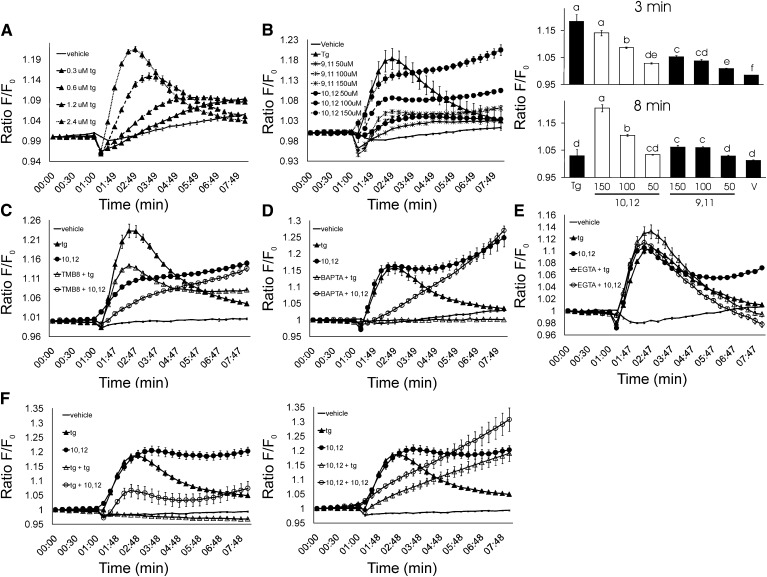
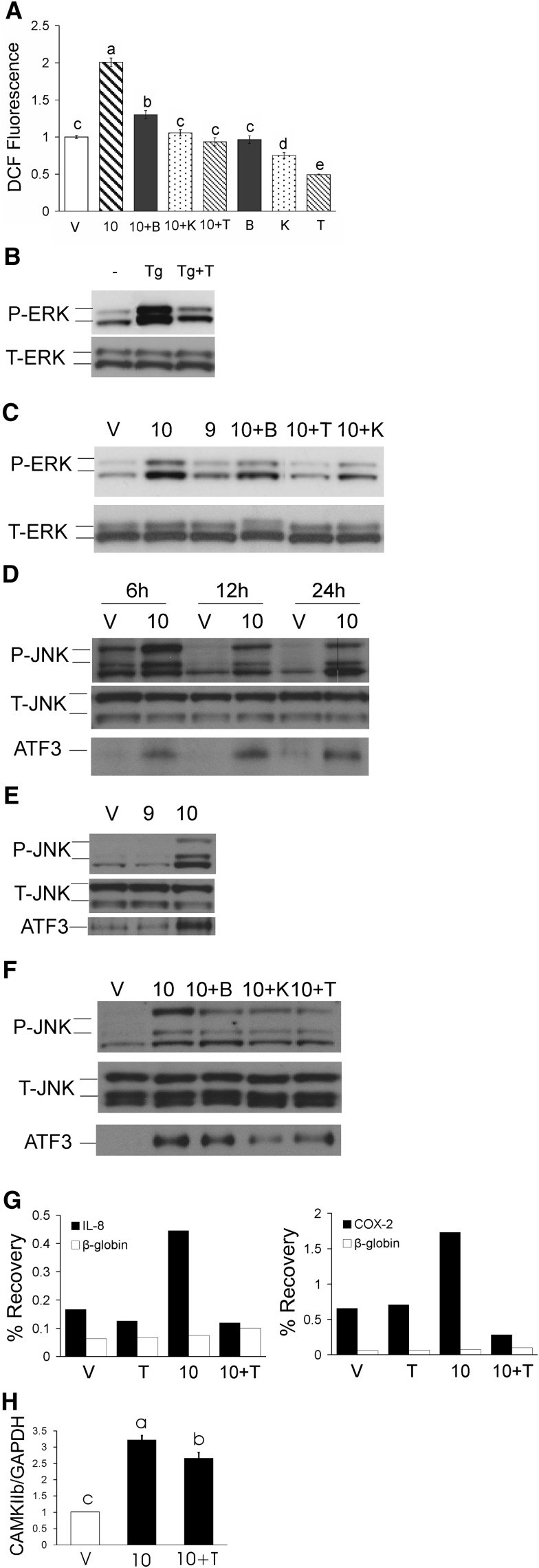
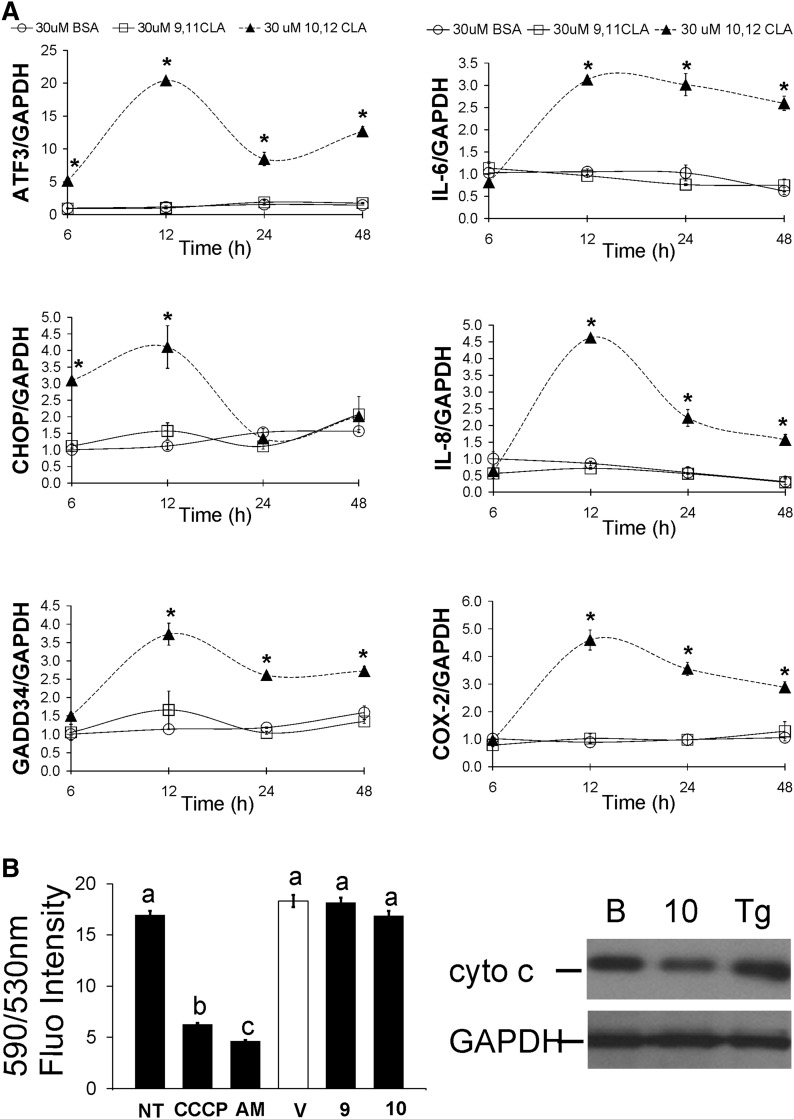
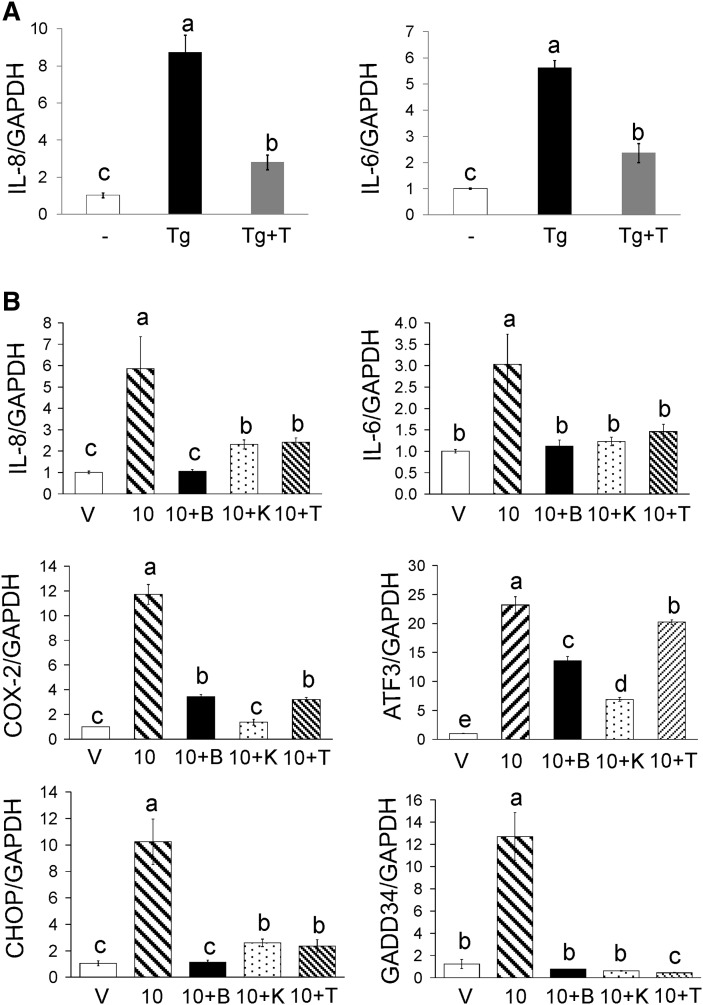
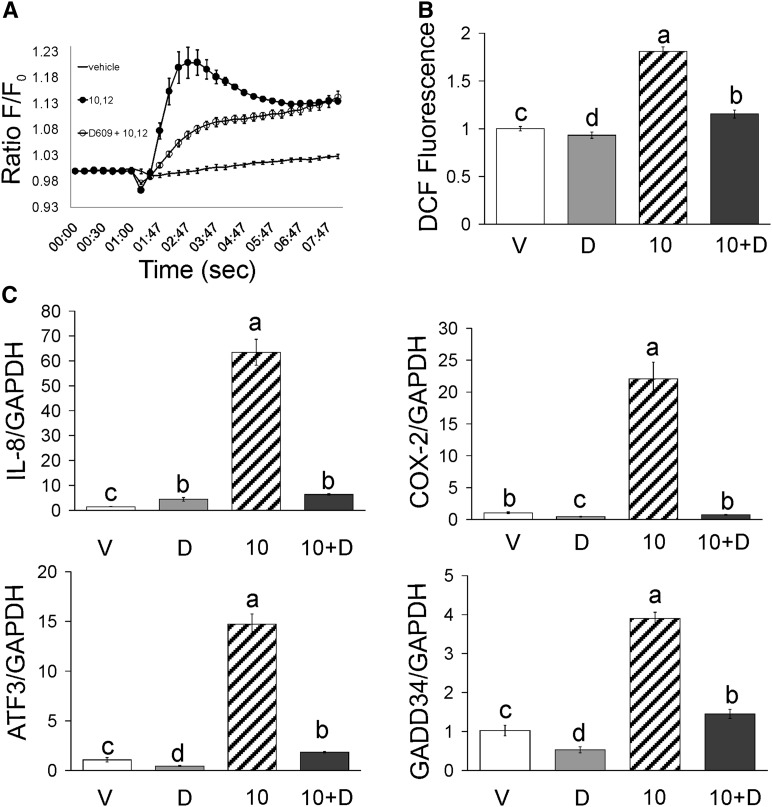
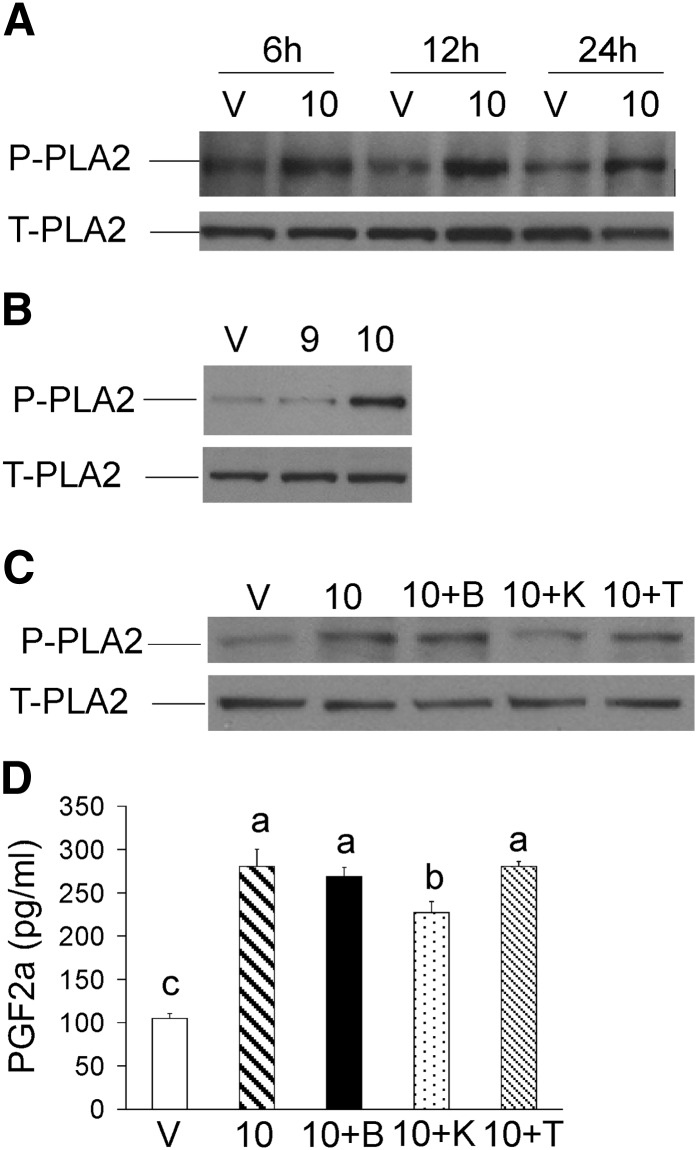
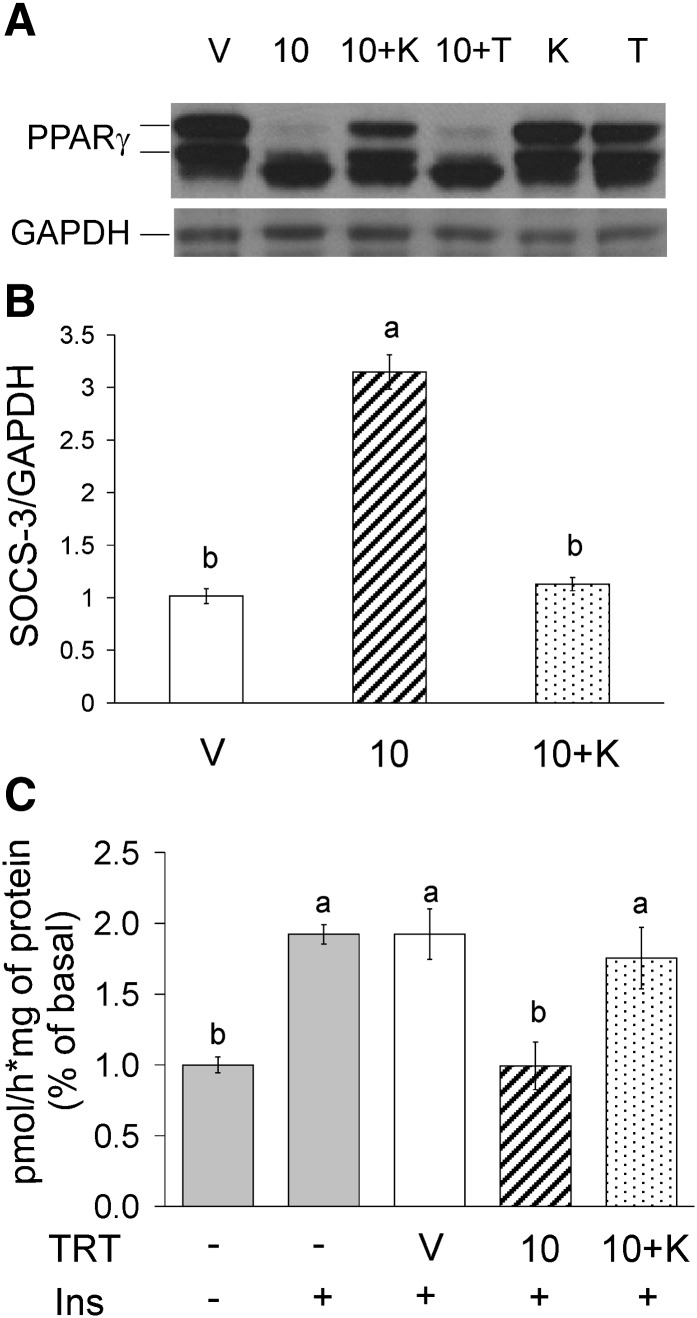
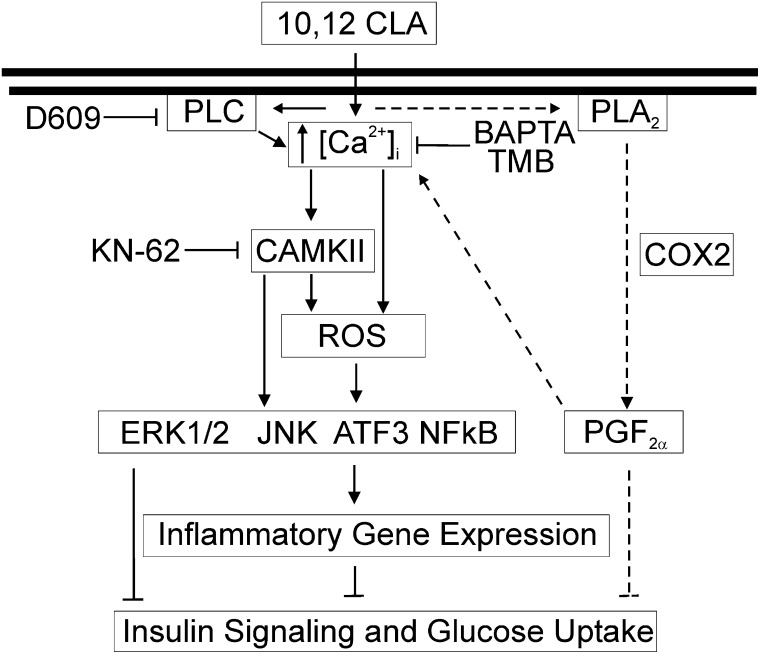
We previously demonstrated that trans-10, cis-12 (10,12) conjugated linoleic acid (CLA) induced inflammation and insulin resistance in primary human adipocytes by activating nuclear factor kappaB (NFkappaB) and extracellular signal-related kinase (ERK) signaling. In this study, we demonstrated that the initial increase in intracellular calcium ([Ca2+]i) mediated by 10,12 CLA was attenuated by TMB-8, an inhibitor of calcium release from the endoplasmic reticulum (ER), by BAPTA, an intracellular calcium chelator, and by D609, a phospholipase C (PLC) inhibitor. Moreover, BAPTA, TMB-8, and D609 attenuated 10,12 CLA-mediated production of reactive oxygen species (ROS), activation of ERK1/2 and cJun-NH2-terminal kinase (JNK), and induction of inflammatory genes. 10,12 CLA-mediated binding of NFkappaB to the promoters of interleukin (IL)-8 and cyclooxygenase (COX)-2 and induction of calcium-calmodulin kinase II (CaMKII) beta were attenuated by TMB-8. KN-62, a CaMKII inhibitor, also suppressed 10,12 CLA-mediated ROS production and ERK1/2 and JNK activation. Additionally, KN-62 attenuated 10,12 CLA induction of inflammatory and integrated stress response genes, increase in prostaglandin F2alpha, and suppression of peroxisome proliferator activated receptor gamma protein levels and insulin-stimulated glucose uptake. These data suggest that 10,12 CLA increases inflammation and insulin resistance in human adipocytes, in part by increasing [Ca2+]i levels, particularly calcium from the ER.
Figures
References
-
- Centers for Disease Control and Prevention. US Obesity Trends, 1985–2006. Accessed 2007 at http://www.cdc.gov/nccdphp/dnpa/obesity/trend/maps/index.htm.
-
- Whigham L. D., Watras A. C., Schoeller D. A. 2007. Efficacy of conjugated linoleic acid for reducing fat mass: a meta-analysis in humans. Am. J. Clin. Nutr. 85: 1203–1211. - PubMed
-
- Granlund L., Juvet L. K., Pedersen J. I., Nebb H. I. 2003. Trans10, cis12-conjugated linoleic acid prevents triacylglycerol accumulation in adipocytes by acting as a PPARgamma modulator. J. Lipid Res. 44: 1441–1452. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
