

Voltage-sensor mutations in channelopathies of skeletal muscle
- PMID: 20156847
- PMCID: PMC2901977
- DOI: 10.1113/jphysiol.2010.186874
Voltage-sensor mutations in channelopathies of skeletal muscle
Abstract
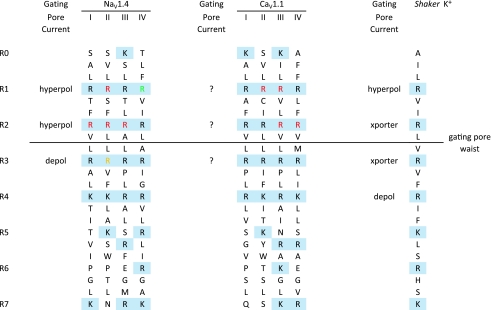
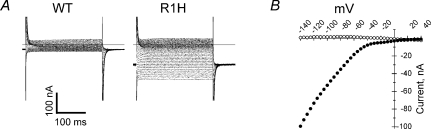
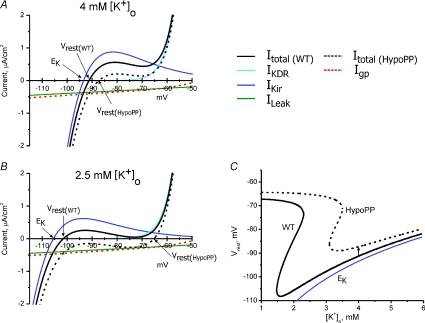
Mutations of voltage-gated ion channels cause several channelopathies of skeletal muscle, which present clinically with myotonia, periodic paralysis, or a combination of both. Expression studies have revealed both loss-of-function and gain-of-function defects for the currents passed by mutant channels. In many cases, these functional changes could be mechanistically linked to the defects of fibre excitability underlying myotonia or periodic paralysis. One remaining enigma was the basis for depolarization-induced weakness in hypokalaemic periodic paralysis (HypoPP) arising from mutations in either sodium or calcium channels. Curiously, 14 of 15 HypoPP mutations are at arginines in S4 voltage sensors, and recent observations show that these substitutions support an alternative pathway for ion conduction, the gating pore, that may be the source of the aberrant depolarization during an attack of paralysis.
Figures
References
-
- Ashcroft FM. Ion Channels and Disease. Oxford: Elsevier; 2000.
-
- Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, Ebers GC. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. 1999;53:1932–1936. - PubMed
-
- Cannon SC. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci. 2006;29:387–415. - PubMed
-
- Cannon SC, Brown RH, Jr, Corey DP. A sodium channel defect in hyperkalemic periodic paralysis: potassium-induced failure of inactivation. Neuron. 1991;6:619–626. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous