

Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein
- PMID: 20156965
- PMCID: PMC2828915
- DOI: 10.1083/jcb.200911115
Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein
Abstract
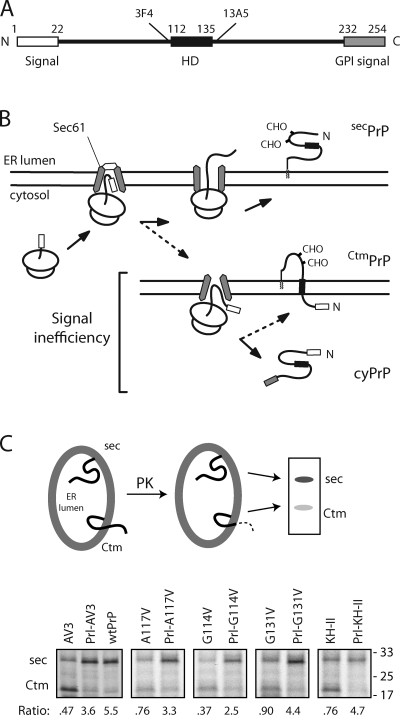
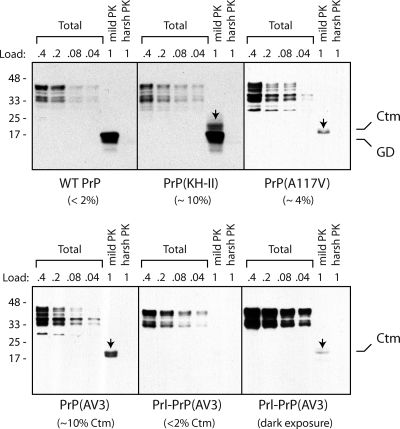
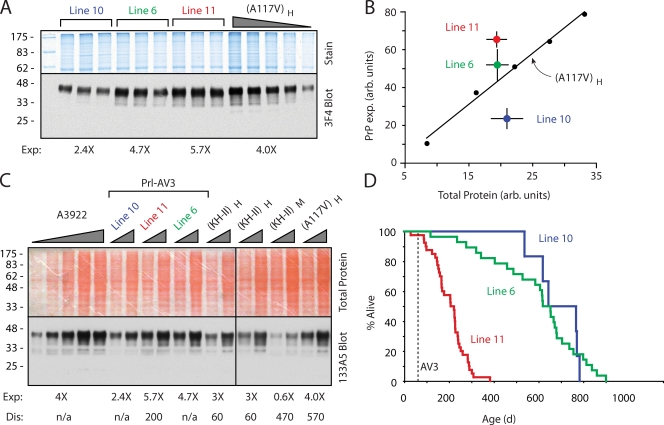
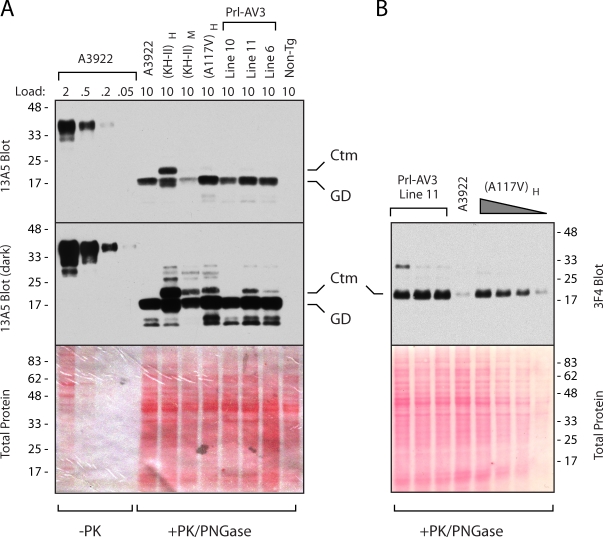
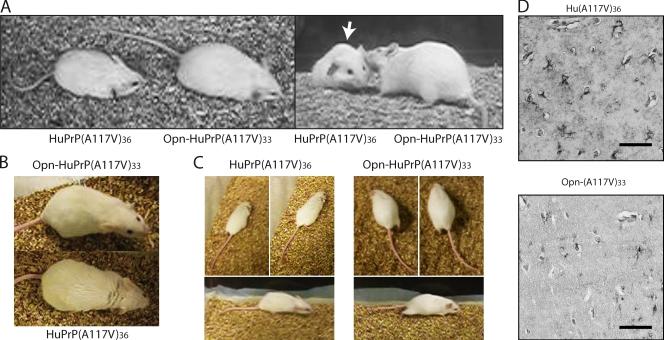
Protein translocation into the endoplasmic reticulum is mediated by signal sequences that vary widely in primary structure. In vitro studies suggest that such signal sequence variations may correspond to subtly different functional properties. Whether comparable functional differences exist in vivo and are of sufficient magnitude to impact organism physiology is unknown. Here, we investigate this issue by analyzing in transgenic mice the impact of signal sequence efficiency for mammalian prion protein (PrP). We find that replacement of the average efficiency signal sequence of PrP with more efficient signals rescues mice from neurodegeneration caused by otherwise pathogenic PrP mutants in a downstream hydrophobic domain (HD). This effect is explained by the demonstration that efficient signal sequence function precludes generation of a cytosolically exposed, disease-causing transmembrane form of PrP mediated by the HD mutants. Thus, signal sequences are functionally nonequivalent in vivo, with intrinsic inefficiency of the native PrP signal being required for pathogenesis of a subset of disease-causing PrP mutations.
Figures

Similar articles
-
Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration.Dev Cell. 2008 Sep;15(3):359-370. doi: 10.1016/j.devcel.2008.06.015. Dev Cell. 2008. PMID: 18804434 Free PMC article.
-
A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic Reticulum.Mol Biol Cell. 2001 Apr;12(4):881-9. doi: 10.1091/mbc.12.4.881. Mol Biol Cell. 2001. PMID: 11294893 Free PMC article.
-
Mutational analysis of topological determinants in prion protein (PrP) and measurement of transmembrane and cytosolic PrP during prion infection.J Biol Chem. 2003 Nov 14;278(46):45960-8. doi: 10.1074/jbc.M307833200. Epub 2003 Aug 21. J Biol Chem. 2003. PMID: 12933795
-
Analyzing the influence of PrP primary structure on prion pathogenesis in transgenic mice.Arch Virol Suppl. 2000;(16):87-94. doi: 10.1007/978-3-7091-6308-5_7. Arch Virol Suppl. 2000. PMID: 11214937 Review.
-
The role of PrP in health and disease.Curr Mol Med. 2004 Jun;4(4):337-53. doi: 10.2174/1566524043360645. Curr Mol Med. 2004. PMID: 15354865 Review.
Cited by
-
INS-gene mutations: from genetics and beta cell biology to clinical disease.Mol Aspects Med. 2015 Apr;42:3-18. doi: 10.1016/j.mam.2014.12.001. Epub 2014 Dec 24. Mol Aspects Med. 2015. PMID: 25542748 Free PMC article. Review.
-
PrP-containing aggresomes are cytosolic components of an ER quality control mechanism.J Cell Sci. 2016 Oct 1;129(19):3635-3647. doi: 10.1242/jcs.186981. Epub 2016 Aug 22. J Cell Sci. 2016. PMID: 27550517 Free PMC article.
-
Homodimerization as a molecular switch between low and high efficiency PrP C cell surface delivery and neuroprotective activity.Prion. 2013 Mar-Apr;7(2):170-4. doi: 10.4161/pri.23583. Epub 2013 Jan 28. Prion. 2013. PMID: 23357826 Free PMC article.
-
The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain.Cells. 2020 Mar 2;9(3):591. doi: 10.3390/cells9030591. Cells. 2020. PMID: 32131451 Free PMC article. Review.
-
The C-degron pathway eliminates mislocalized proteins and products of deubiquitinating enzymes.EMBO J. 2021 Apr 1;40(7):e105846. doi: 10.15252/embj.2020105846. Epub 2021 Jan 20. EMBO J. 2021. PMID: 33469951 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
