

Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations
- PMID: 20157008
- PMCID: PMC2842515
- DOI: 10.1093/brain/awq015
Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations
Abstract
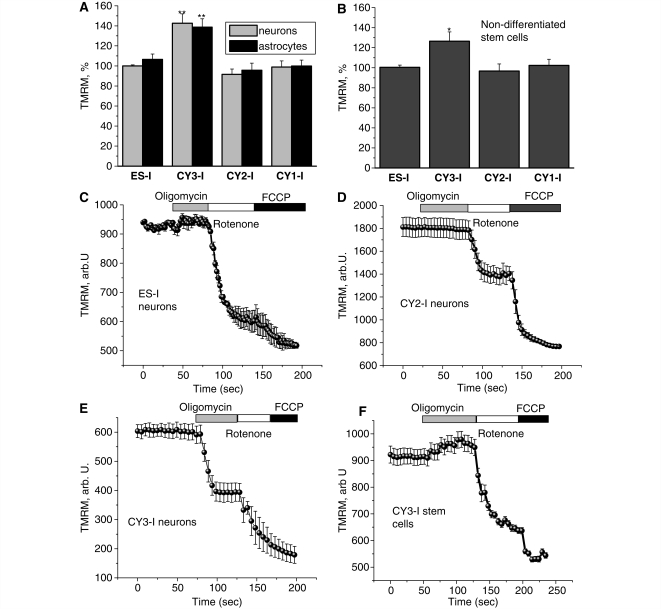
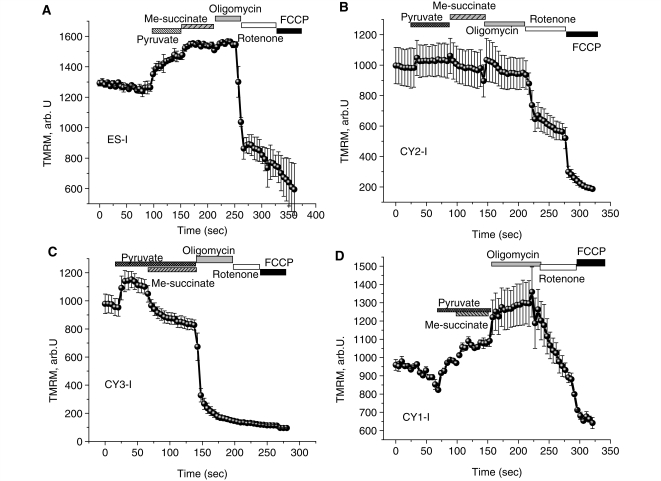
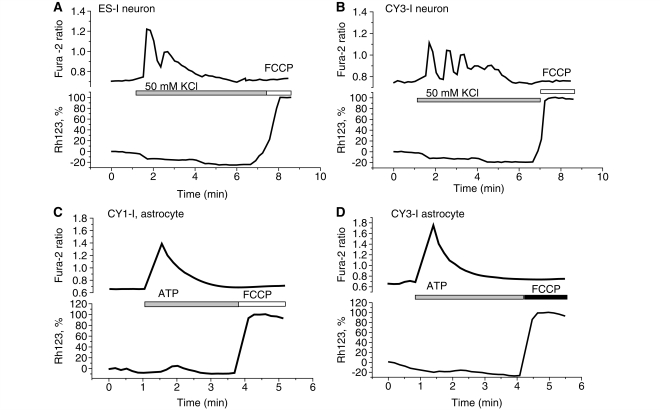
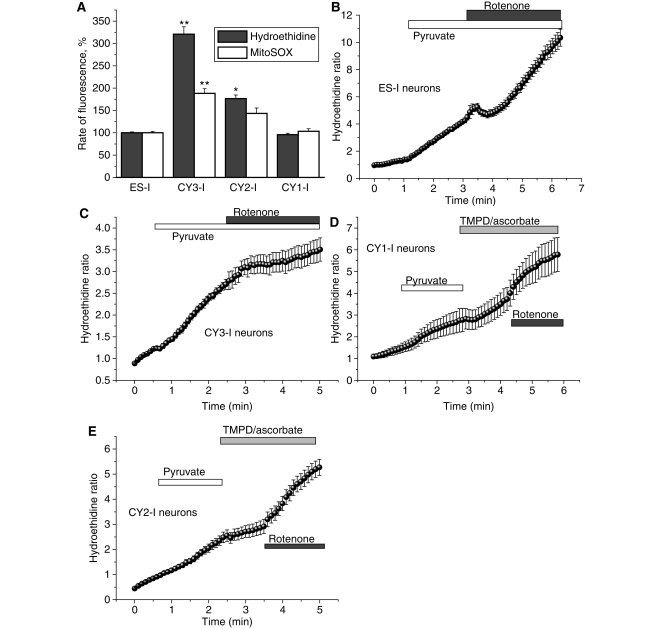
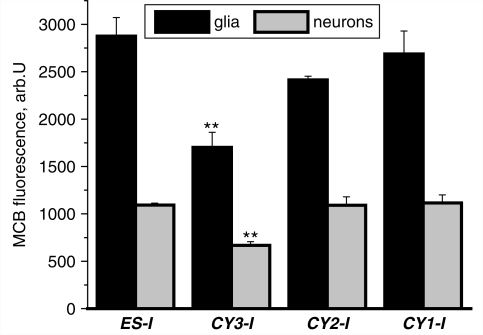
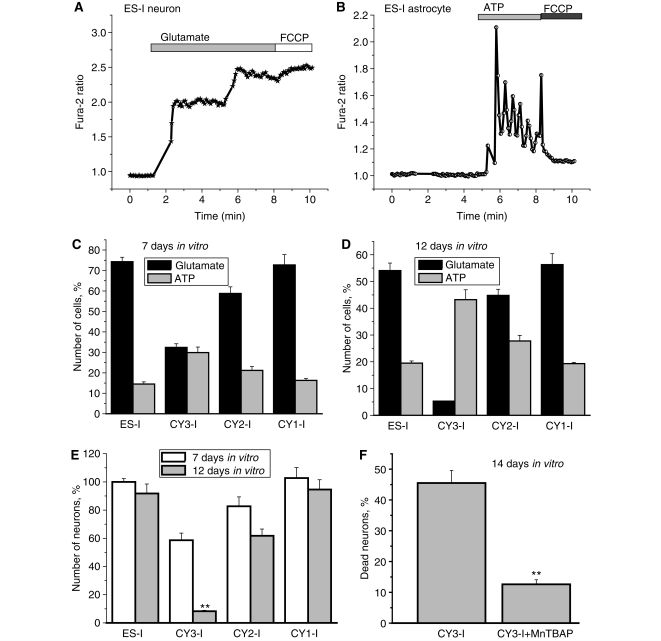
Mutations of mitochondrial DNA are associated with a wide spectrum of disorders, primarily affecting the central nervous system and muscle function. The specific consequences of mitochondrial DNA mutations for neuronal pathophysiology are not understood. In order to explore the impact of mitochondrial mutations on neuronal biochemistry and physiology, we have used fluorescence imaging techniques to examine changes in mitochondrial function in neurons differentiated from mouse embryonic stem-cell cybrids containing mitochondrial DNA polymorphic variants or mutations. Surprisingly, in neurons carrying a severe mutation in respiratory complex I (<10% residual complex I activity) the mitochondrial membrane potential was significantly increased, but collapsed in response to oligomycin, suggesting that the mitochondrial membrane potential was maintained by the F(1)F(o) ATPase operating in 'reverse' mode. In cells with a mutation in complex IV causing approximately 40% residual complex IV activity, the mitochondrial membrane potential was not significantly different from controls. The rate of generation of mitochondrial reactive oxygen species, measured using hydroethidium and signals from the mitochondrially targeted hydroethidine, was increased in neurons with both the complex I and complex IV mutations. Glutathione was depleted, suggesting significant oxidative stress in neurons with a complex I deficiency, but not in those with a complex IV defect. In the neurons with complex I deficiency but not the complex IV defect, neuronal death was increased and was attenuated by reactive oxygen species scavengers. Thus, in neurons with a severe mutation of complex I, the maintenance of a high potential by F(1)F(o) ATPase activity combined with an impaired respiratory chain causes oxidative stress which promotes cell death.
Figures
References
-
- Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta. 2008;1777:953–64. - PubMed
-
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7. - PubMed
-
- Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, et al. Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol. 2008;255:1231–5. - PubMed
-
- Betts J, Jaros E, Perry RH, Schaefer AM, Taylor RW, Abdel-All Z, et al. Molecular neuropathology of MELAS: level of heteroplasmy in individual neurones and evidence of extensive vascular involvement. Neuropathol Appl Neurobiol. 2006;32:359–73. - PubMed
