

Multi-system neurological disease is common in patients with OPA1 mutations
- PMID: 20157015
- PMCID: PMC2842512
- DOI: 10.1093/brain/awq007
Multi-system neurological disease is common in patients with OPA1 mutations
Abstract
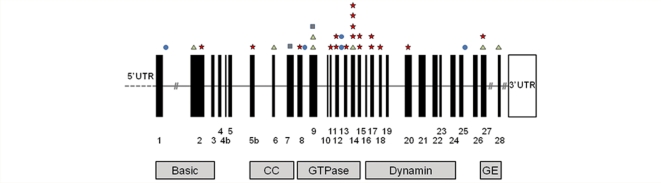
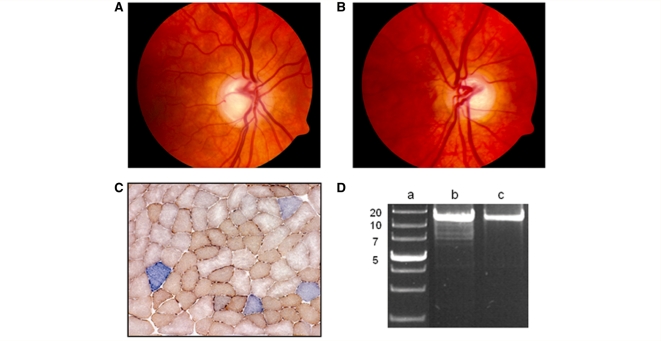
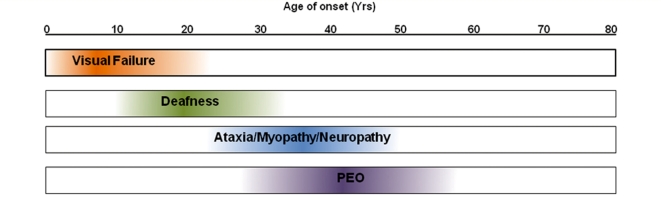
Additional neurological features have recently been described in seven families transmitting pathogenic mutations in OPA1, the most common cause of autosomal dominant optic atrophy. However, the frequency of these syndromal 'dominant optic atrophy plus' variants and the extent of neurological involvement have not been established. In this large multi-centre study of 104 patients from 45 independent families, including 60 new cases, we show that extra-ocular neurological complications are common in OPA1 disease, and affect up to 20% of all mutational carriers. Bilateral sensorineural deafness beginning in late childhood and early adulthood was a prominent manifestation, followed by a combination of ataxia, myopathy, peripheral neuropathy and progressive external ophthalmoplegia from the third decade of life onwards. We also identified novel clinical presentations with spastic paraparesis mimicking hereditary spastic paraplegia, and a multiple sclerosis-like illness. In contrast to initial reports, multi-system neurological disease was associated with all mutational subtypes, although there was an increased risk with missense mutations [odds ratio = 3.06, 95% confidence interval = 1.44-6.49; P = 0.0027], and mutations located within the guanosine triphosphate-ase region (odds ratio = 2.29, 95% confidence interval = 1.08-4.82; P = 0.0271). Histochemical and molecular characterization of skeletal muscle biopsies revealed the presence of cytochrome c oxidase-deficient fibres and multiple mitochondrial DNA deletions in the majority of patients harbouring OPA1 mutations, even in those with isolated optic nerve involvement. However, the cytochrome c oxidase-deficient load was over four times higher in the dominant optic atrophy + group compared to the pure optic neuropathy group, implicating a causal role for these secondary mitochondrial DNA defects in disease pathophysiology. Individuals with dominant optic atrophy plus phenotypes also had significantly worse visual outcomes, and careful surveillance is therefore mandatory to optimize the detection and management of neurological disability in a group of patients who already have significant visual impairment.
Figures





Comment in
-
Heterozygous OPA1 mutations in Behr syndrome.Brain. 2011 Apr;134(Pt 4):e169; author reply e170. doi: 10.1093/brain/awq306. Epub 2010 Nov 26. Brain. 2011. PMID: 21112924 No abstract available.
-
Spastic paraplegia in 'dominant optic atrophy plus' phenotype due to OPA1 mutation.Brain. 2011 Nov;134(Pt 11):e195. doi: 10.1093/brain/awr101. Epub 2011 Jun 6. Brain. 2011. PMID: 21646330 No abstract available.
-
Sensorineural hearing loss in OPA1-linked disorders.Brain. 2013 Jul;136(Pt 7):e236. doi: 10.1093/brain/aws340. Epub 2013 Feb 4. Brain. 2013. PMID: 23384603 No abstract available.
-
Reply: Sensorineural hearing loss in OPA1-linked disorders.Brain. 2013 Jul;136(Pt 7):e237. doi: 10.1093/brain/aws341. Epub 2013 Feb 4. Brain. 2013. PMID: 23650221 Free PMC article. No abstract available.
-
Reply: 'Behr syndrome' with OPA1 compound heterozygote mutations.Brain. 2015 Jan;138(Pt 1):e322. doi: 10.1093/brain/awu235. Epub 2014 Aug 21. Brain. 2015. PMID: 25146915 Free PMC article. No abstract available.
-
'Behr syndrome' with OPA1 compound heterozygote mutations.Brain. 2015 Jan;138(Pt 1):e321. doi: 10.1093/brain/awu234. Epub 2014 Aug 21. Brain. 2015. PMID: 25146916 Free PMC article. No abstract available.
References
-
- Alexander C, Votruba M, Pesch UEA, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–5. - PubMed
-
- Amati-Bonneau P, Guichet A, Olichon A, Chevrollier A, Viala F, Miot S, et al. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Annals of Neurology. 2005;58:958–63. - PubMed
-
- Amati-Bonneau P, Pasquier L, Lainey E, Ferre M, Odent S, Malthiery Y, et al. Sporadic optic atrophy due to synonymous codon change altering mRNA splicing of OPA1. Clin Genet. 2004;67:102–3. - PubMed
-
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy plus phenotypes. Brain. 2008;131:338–51. - PubMed
-
- Bigal ME, Liberman JN, Lipton RB. Age-dependent prevalence and clinical features of migraine. Neurology. 2006;67:246–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
