

Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1
- PMID: 20157049
- PMCID: PMC3080599
- DOI: 10.1161/HYPERTENSIONAHA.109.149542
Pentaerythritol tetranitrate improves angiotensin II-induced vascular dysfunction via induction of heme oxygenase-1
Abstract
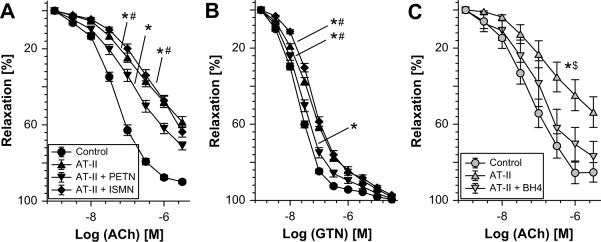
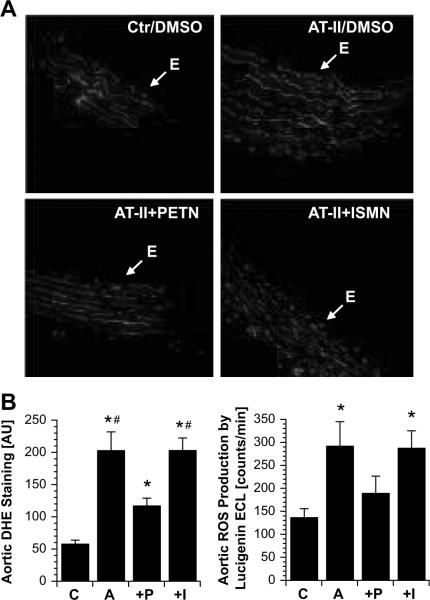
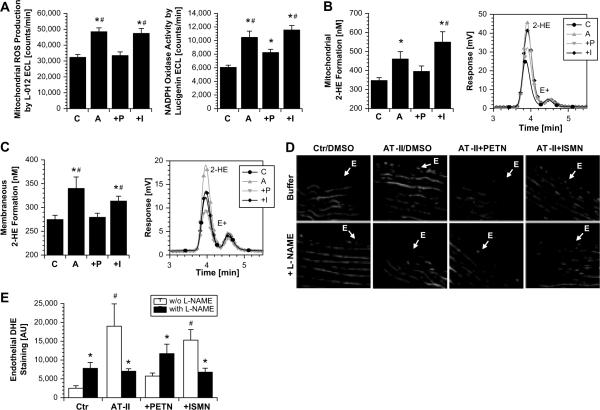
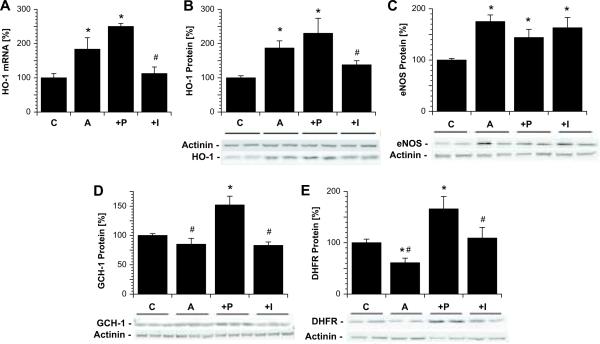
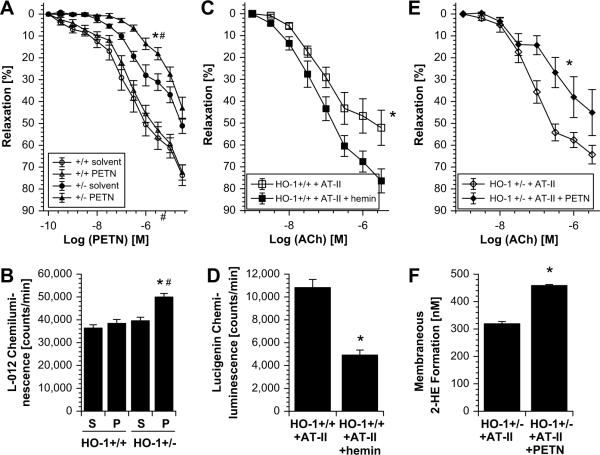
The organic nitrate pentaerythritol tetranitrate is devoid of nitrate tolerance, which has been attributed to the induction of the antioxidant enzyme heme oxygenase (HO)-1. With the present study, we tested whether chronic treatment with pentaerythritol tetranitrate can improve angiotensin II-induced vascular oxidative stress and dysfunction. In contrast to isosorbide-5 mononitrate (75 mg/kg per day for 7 days), treatment with pentaerythritol tetranitrate (15 mg/kg per day for 7 days) improved the impaired endothelial and smooth muscle function and normalized vascular and cardiac reactive oxygen species production (mitochondria, NADPH oxidase activity, and uncoupled endothelial NO synthase), as assessed by dihydroethidine staining, lucigenin-enhanced chemiluminescence, and quantification of dihydroethidine oxidation products in angiotensin II (1 mg/kg per day for 7 days)-treated rats. The antioxidant features of pentaerythritol tetranitrate were recapitulated in spontaneously hypertensive rats. In addition to an increase in HO-1 protein expression, pentaerythritol tetranitrate but not isosorbide-5 mononitrate normalized vascular reactive oxygen species formation and augmented aortic protein levels of the tetrahydrobiopterin-synthesizing enzymes GTP-cyclohydrolase I and dihydrofolate reductase in angiotensin II-treated rats, thereby preventing endothelial NO synthase uncoupling. Haploinsufficiency of HO-1 completely abolished the beneficial effects of pentaerythritol tetranitrate in angiotensin II-treated mice, whereas HO-1 induction by hemin (25 mg/kg) mimicked the effect of pentaerythritol tetranitrate. Improvement of vascular function in this particular model of arterial hypertension by pentaerythritol tetranitrate largely depends on the induction of the antioxidant enzyme HO-1 and identifies pentaerythritol tetranitrate, in contrast to isosorbide-5 mononitrate, as an organic nitrate able to improve rather than to worsen endothelial function.
Figures
Similar articles
-
Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy.Diabetes. 2011 Oct;60(10):2608-16. doi: 10.2337/db10-1395. Epub 2011 Aug 15. Diabetes. 2011. PMID: 21844097 Free PMC article.
-
Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring.Hypertension. 2015 Jan;65(1):232-7. doi: 10.1161/HYPERTENSIONAHA.114.04416. Epub 2014 Nov 10. Hypertension. 2015. PMID: 25385760
-
Pentaerythritol Tetranitrate Targeting Myocardial Reactive Oxygen Species Production Improves Left Ventricular Remodeling and Function in Rats With Ischemic Heart Failure.Hypertension. 2015 Nov;66(5):978-87. doi: 10.1161/HYPERTENSIONAHA.115.05931. Epub 2015 Sep 8. Hypertension. 2015. PMID: 26351025
-
Non-hemodynamic effects of organic nitrates and the distinctive characteristics of pentaerithrityl tetranitrate.Am J Cardiovasc Drugs. 2009;9(1):7-15. doi: 10.1007/BF03256591. Am J Cardiovasc Drugs. 2009. PMID: 19178128 Review.
-
Organic Nitrate Therapy, Nitrate Tolerance, and Nitrate-Induced Endothelial Dysfunction: Emphasis on Redox Biology and Oxidative Stress.Antioxid Redox Signal. 2015 Oct 10;23(11):899-942. doi: 10.1089/ars.2015.6376. Epub 2015 Sep 24. Antioxid Redox Signal. 2015. PMID: 26261901 Free PMC article. Review.
Cited by
-
Fetal programming effects of pentaerythritol tetranitrate in a rat model of superimposed preeclampsia.J Mol Med (Berl). 2020 Sep;98(9):1287-1299. doi: 10.1007/s00109-020-01949-0. Epub 2020 Aug 3. J Mol Med (Berl). 2020. PMID: 32748067 Free PMC article.
-
Organic nitrates and nitrate resistance in diabetes: the role of vascular dysfunction and oxidative stress with emphasis on antioxidant properties of pentaerithrityl tetranitrate.Exp Diabetes Res. 2010;2010:213176. doi: 10.1155/2010/213176. Epub 2010 Dec 27. Exp Diabetes Res. 2010. PMID: 21234399 Free PMC article. Review.
-
Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system.Redox Biol. 2017 Aug;12:35-49. doi: 10.1016/j.redox.2017.02.001. Epub 2017 Feb 7. Redox Biol. 2017. PMID: 28212522 Free PMC article. Review.
-
Renal Effects of Fetal Reprogramming With Pentaerythritol Tetranitrate in Spontaneously Hypertensive Rats.Front Pharmacol. 2020 Apr 29;11:454. doi: 10.3389/fphar.2020.00454. eCollection 2020. Front Pharmacol. 2020. PMID: 32410988 Free PMC article.
-
Noise-Induced Vascular Dysfunction, Oxidative Stress, and Inflammation Are Improved by Pharmacological Modulation of the NRF2/HO-1 Axis.Antioxidants (Basel). 2021 Apr 19;10(4):625. doi: 10.3390/antiox10040625. Antioxidants (Basel). 2021. PMID: 33921821 Free PMC article.
References
-
- Hamilton C. Nitric oxide, oxidative stress and hypertension: A complex equation. J Hypertens. 2002;20:1055–1056. - PubMed
-
- Touyz RM. Reactive oxygen species in vascular biology: Role in arterial hypertension. Expert Rev Cardiovasc Ther. 2003;1:91–106. - PubMed
-
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin ii-mediated hypertension in the rat increases vascular superoxide production via membrane nadh/nadph oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. - PMC - PubMed
-
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin ii infusion on the expression and function of nad(p)h oxidase and components of nitric oxide/cgmp signaling. Circ Res. 2002;90:E58–65. - PubMed
-
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin ii-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
