

Analysis of CASP8 targets, predictions and assessment methods
- PMID: 20157476
- PMCID: PMC2794793
- DOI: 10.1093/database/bap003
Analysis of CASP8 targets, predictions and assessment methods
Abstract
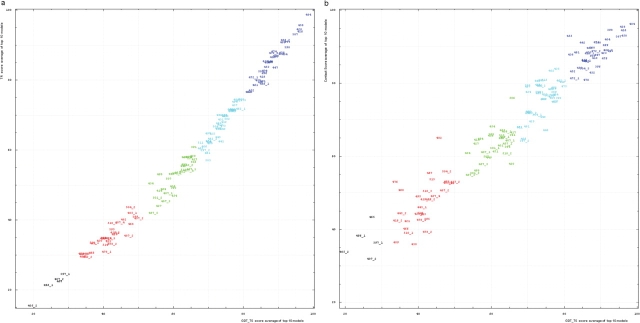
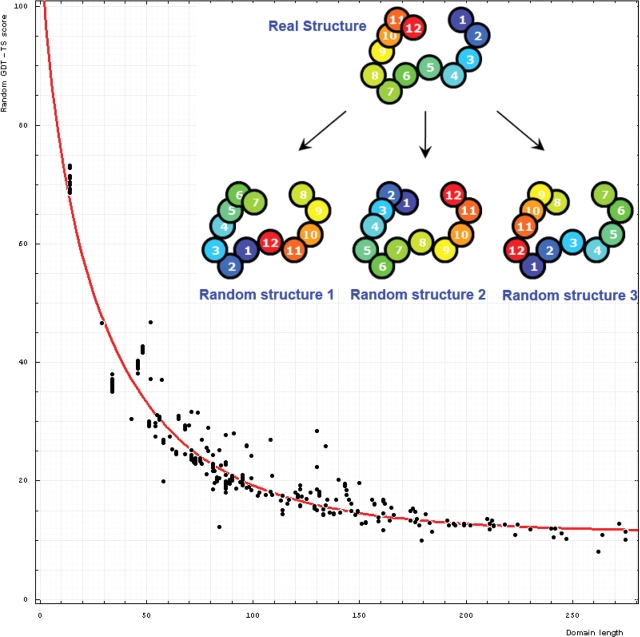
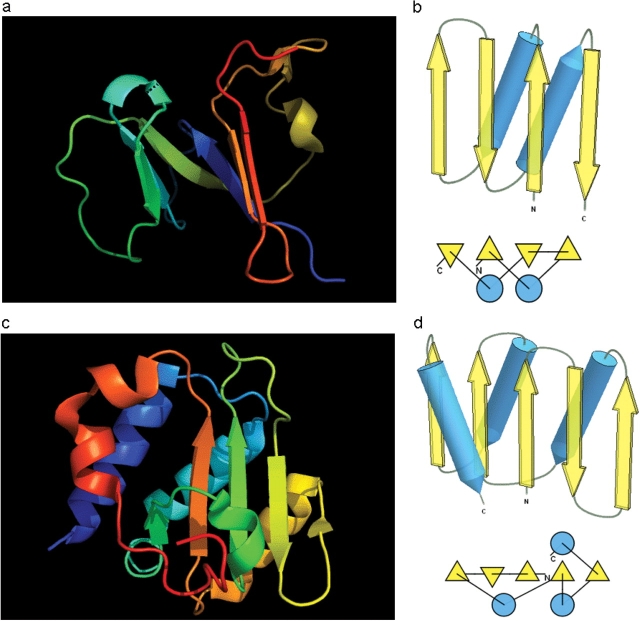
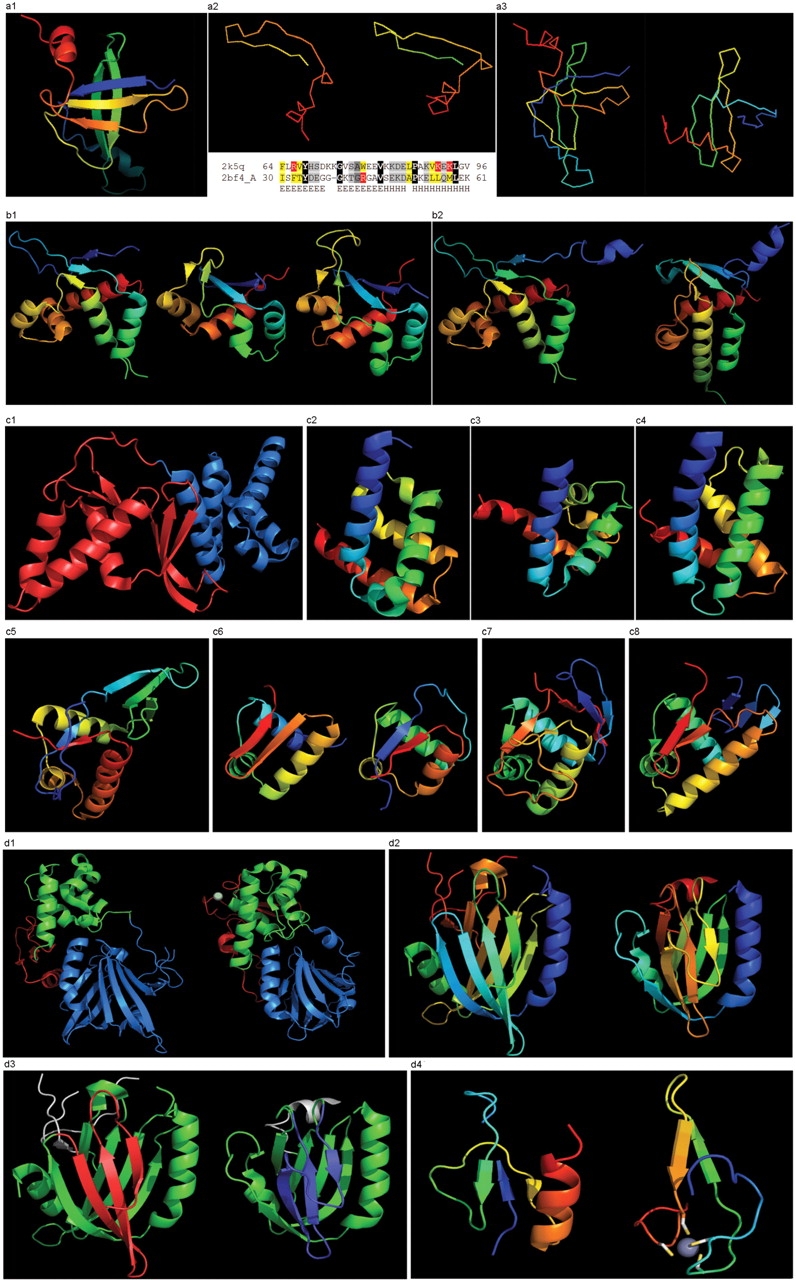
Results of the recent Critical Assessment of Techniques for Protein Structure Prediction, CASP8, present several valuable sources of information. First, CASP targets comprise a realistic sample of currently solved protein structures and exemplify the corresponding challenges for predictors. Second, the plethora of predictions by all possible methods provides an unusually rich material for evolutionary analysis of target proteins. Third, CASP results show the current state of the field and highlight specific problems in both predicting and assessing. Finally, these data can serve as grounds to develop and analyze methods for assessing prediction quality. Here we present results of our analysis in these areas. Our objective is not to duplicate CASP assessment, but to use our unique experience as former CASP5 assessors and CASP8 predictors to (i) offer more insights into CASP targets and predictions based on expert analysis, including invaluable analysis prior to target structure release; and (ii) develop an assessment methodology tailored towards current challenges in the field. Specifically, we discuss preparing target structures for assessment, parsing protein domains, balancing evaluations based on domains and on whole chains, dividing targets into categories and developing new evaluation scores. We also present evolutionary analysis of the most interesting and challenging targets.Database URL: Our results are available as a comprehensive database of targets and predictions at http://prodata.swmed.edu/CASP8.
Figures
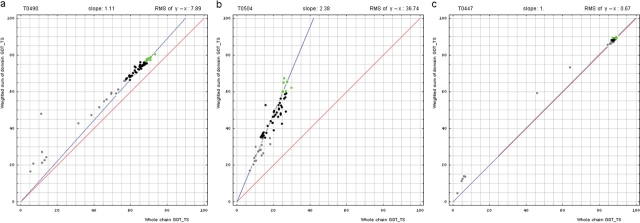
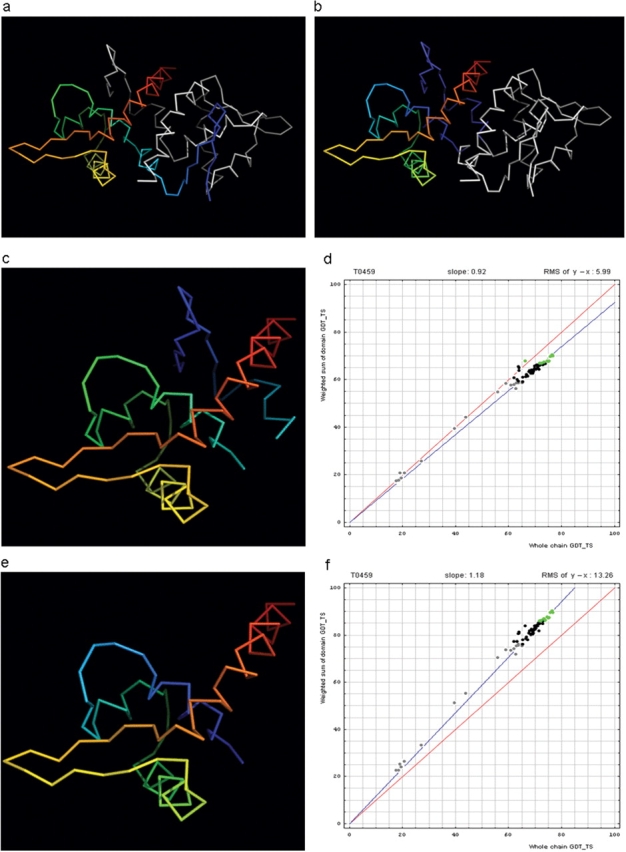
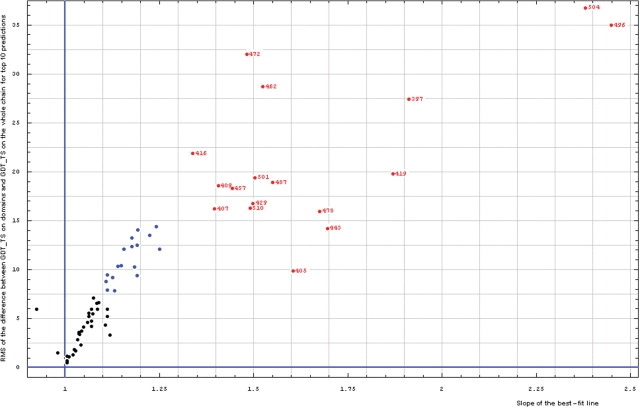
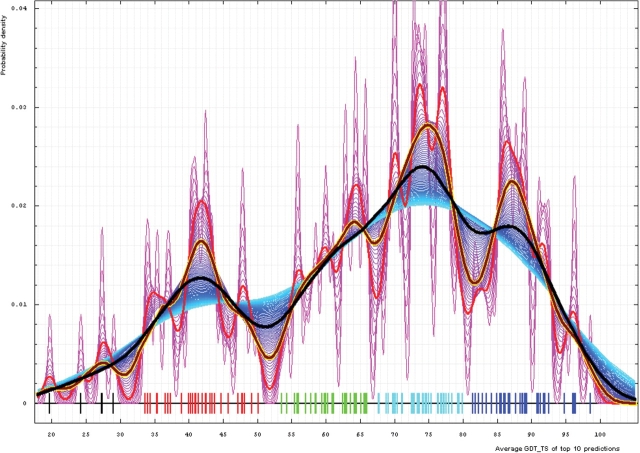
Similar articles
-
CAFASP2: the second critical assessment of fully automated structure prediction methods.Proteins. 2001;Suppl 5:171-83. doi: 10.1002/prot.10036. Proteins. 2001. PMID: 11835495
-
COMPASS server for homology detection: improved statistical accuracy, speed and functionality.Nucleic Acids Res. 2009 Jul;37(Web Server issue):W90-4. doi: 10.1093/nar/gkp360. Epub 2009 May 12. Nucleic Acids Res. 2009. PMID: 19435884 Free PMC article.
-
Assessment of domain boundary predictions and the prediction of intramolecular contacts in CASP8.Proteins. 2009;77 Suppl 9:196-209. doi: 10.1002/prot.22554. Proteins. 2009. PMID: 19714769
-
[Aiming for zero blindness].Nippon Ganka Gakkai Zasshi. 2015 Mar;119(3):168-93; discussion 194. Nippon Ganka Gakkai Zasshi. 2015. PMID: 25854109 Review. Japanese.
-
Review and comparative assessment of similarity-based methods for prediction of drug-protein interactions in the druggable human proteome.Brief Bioinform. 2019 Nov 27;20(6):2066-2087. doi: 10.1093/bib/bby069. Brief Bioinform. 2019. PMID: 30102367 Review.
Cited by
-
Fragment-free approach to protein folding using conditional neural fields.Bioinformatics. 2010 Jun 15;26(12):i310-7. doi: 10.1093/bioinformatics/btq193. Bioinformatics. 2010. PMID: 20529922 Free PMC article.
-
An automatic method for CASP9 free modeling structure prediction assessment.Bioinformatics. 2011 Dec 15;27(24):3371-8. doi: 10.1093/bioinformatics/btr572. Epub 2011 Oct 12. Bioinformatics. 2011. PMID: 21994223 Free PMC article.
-
Databases and bioinformatics tools for the study of DNA repair.Mol Biol Int. 2011;2011:475718. doi: 10.4061/2011/475718. Epub 2011 Jul 14. Mol Biol Int. 2011. PMID: 22091405 Free PMC article.
-
CASP9 target classification.Proteins. 2011;79 Suppl 10(Suppl 10):21-36. doi: 10.1002/prot.23190. Epub 2011 Oct 14. Proteins. 2011. PMID: 21997778 Free PMC article.
-
Structure similarity measure with penalty for close non-equivalent residues.Bioinformatics. 2009 May 15;25(10):1259-63. doi: 10.1093/bioinformatics/btp148. Epub 2009 Mar 25. Bioinformatics. 2009. PMID: 19321733 Free PMC article.
References
-
- Moult J, Pedersen JT, Judson R, et al. A large-scale experiment to assess protein structure prediction methods. Proteins. 1995;23:ii–v. - PubMed
-
- Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 2005;15:285–289. - PubMed
-
- Shammas C, Menne TF, Hilcenko C, et al. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J. Biol. Chem. 2005;280:19221–19229. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
