

MicroScope: a platform for microbial genome annotation and comparative genomics
- PMID: 20157493
- PMCID: PMC2790312
- DOI: 10.1093/database/bap021
MicroScope: a platform for microbial genome annotation and comparative genomics
Abstract
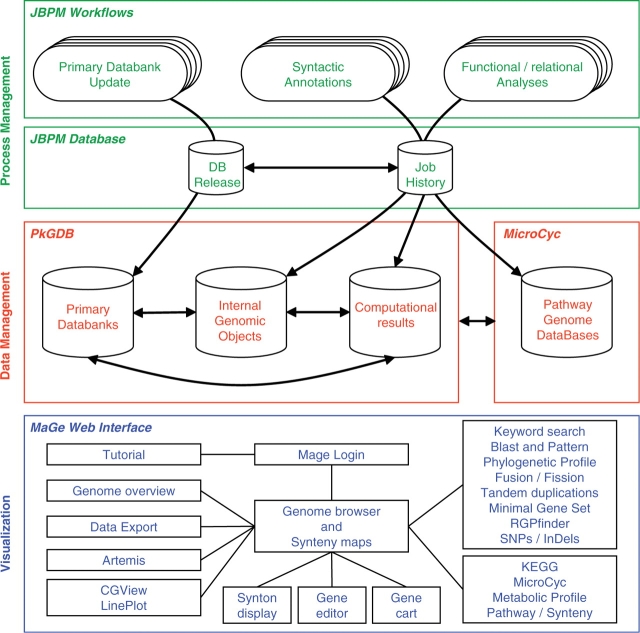
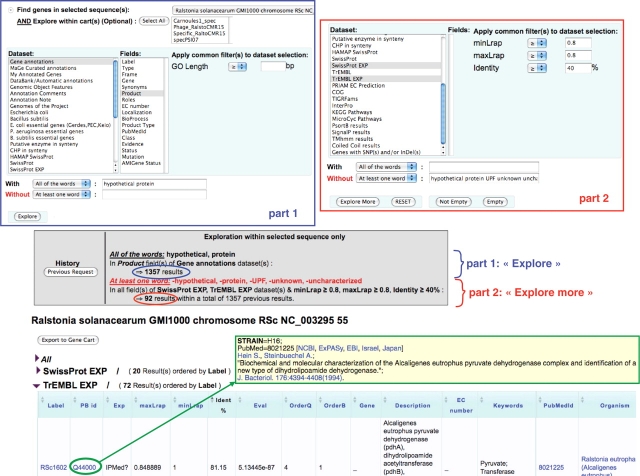
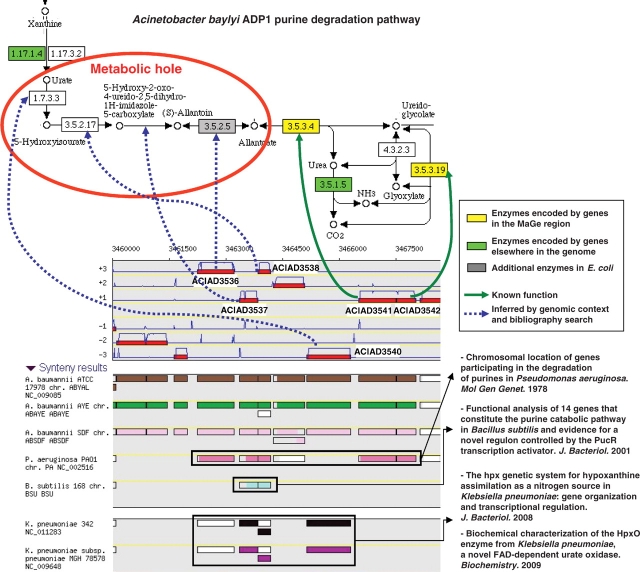
The initial outcome of genome sequencing is the creation of long text strings written in a four letter alphabet. The role of in silico sequence analysis is to assist biologists in the act of associating biological knowledge with these sequences, allowing investigators to make inferences and predictions that can be tested experimentally. A wide variety of software is available to the scientific community, and can be used to identify genomic objects, before predicting their biological functions. However, only a limited number of biologically interesting features can be revealed from an isolated sequence. Comparative genomics tools, on the other hand, by bringing together the information contained in numerous genomes simultaneously, allow annotators to make inferences based on the idea that evolution and natural selection are central to the definition of all biological processes. We have developed the MicroScope platform in order to offer a web-based framework for the systematic and efficient revision of microbial genome annotation and comparative analysis (http://www.genoscope.cns.fr/agc/microscope). Starting with the description of the flow chart of the annotation processes implemented in the MicroScope pipeline, and the development of traditional and novel microbial annotation and comparative analysis tools, this article emphasizes the essential role of expert annotation as a complement of automatic annotation. Several examples illustrate the use of implemented tools for the review and curation of annotations of both new and publicly available microbial genomes within MicroScope's rich integrated genome framework. The platform is used as a viewer in order to browse updated annotation information of available microbial genomes (more than 440 organisms to date), and in the context of new annotation projects (117 bacterial genomes). The human expertise gathered in the MicroScope database (about 280,000 independent annotations) contributes to improve the quality of microbial genome annotation, especially for genomes initially analyzed by automatic procedures alone.Database URLs: http://www.genoscope.cns.fr/agc/mage and http://www.genoscope.cns.fr/agc/microcyc.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
