

A null mutation in CABP4 causes Leber's congenital amaurosis-like phenotype
- PMID: 20157620
- PMCID: PMC2820108
A null mutation in CABP4 causes Leber's congenital amaurosis-like phenotype
Abstract
Purpose: To describe the finding of a novel calcium binding protein 4 (CABP4) mutation in a family with Leber congenital amaurosis (LCA) phenotype.
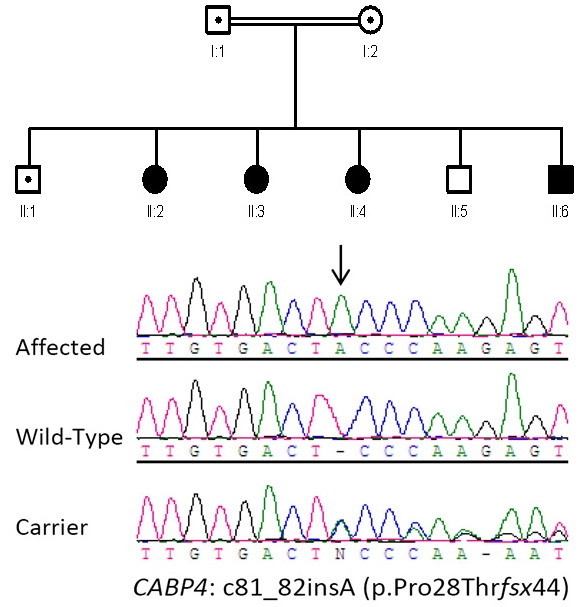
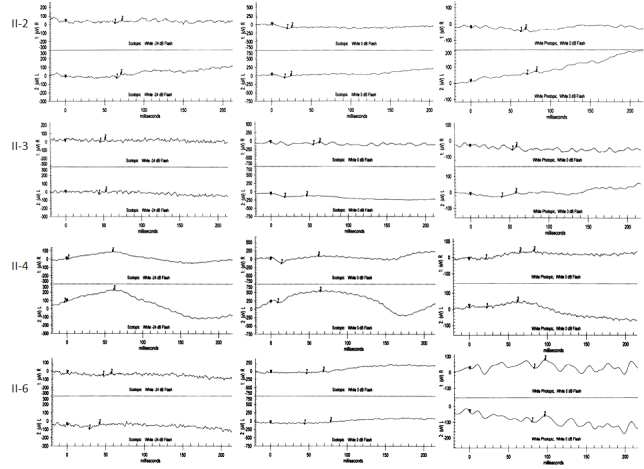
Methods: Homozygosity mapping was performed in a consanguineous family with four affected members originally referred as cases of LCA. Detailed electroretinographic recordings were obtained.
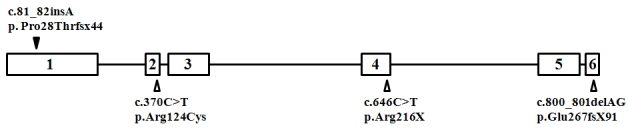
Results: A novel homozygous single base-pair insertion was identified in all four siblings. The patients had an LCA-like phenotype, including either flat or greatly diminished electroretinographic activity.
Conclusions: This report significantly expands on the phenotype associated with calcium binding protein 4 mutations, which has so far been limited to congenital stationary night blindness, and further demonstrates how molecular data often blur the boundaries between what are believed to be clinically distinct retinal disorders.
Figures
References
-
- Dryja TP, Berson EL, Rao VR, Oprian DD. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet. 1993;4:280–3. - PubMed
-
- Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000;130:547–63. - PubMed
-
- Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet. 1994;7:64–8. - PubMed
-
- Yamamoto S, Sippel KC, Berson EL, Dryja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet. 1997;15:175–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials