

Allelic skewing of DNA methylation is widespread across the genome
- PMID: 20159110
- PMCID: PMC2820163
- DOI: 10.1016/j.ajhg.2010.01.014
Allelic skewing of DNA methylation is widespread across the genome
Abstract
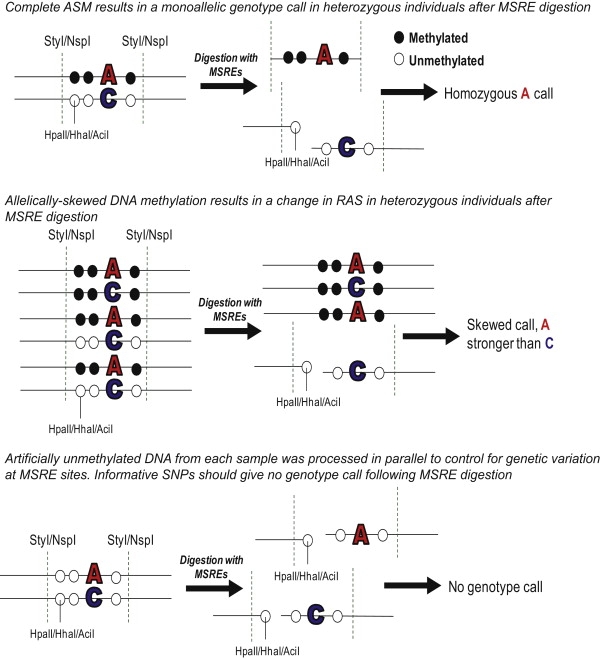
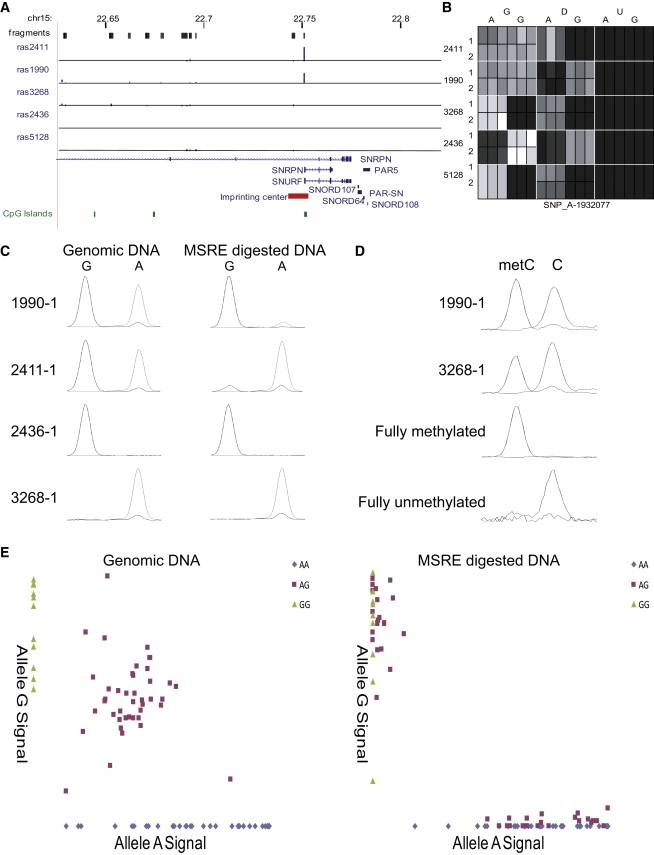
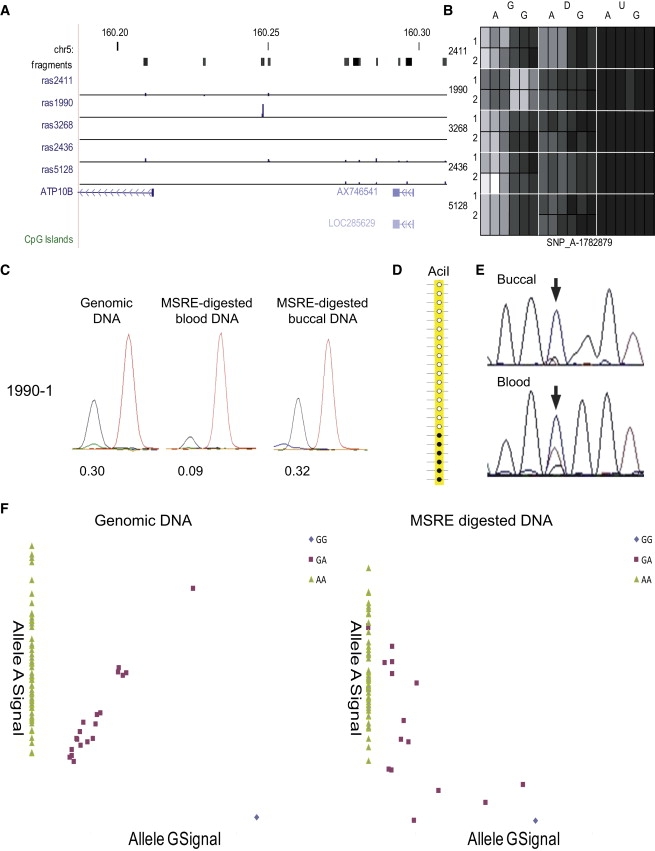
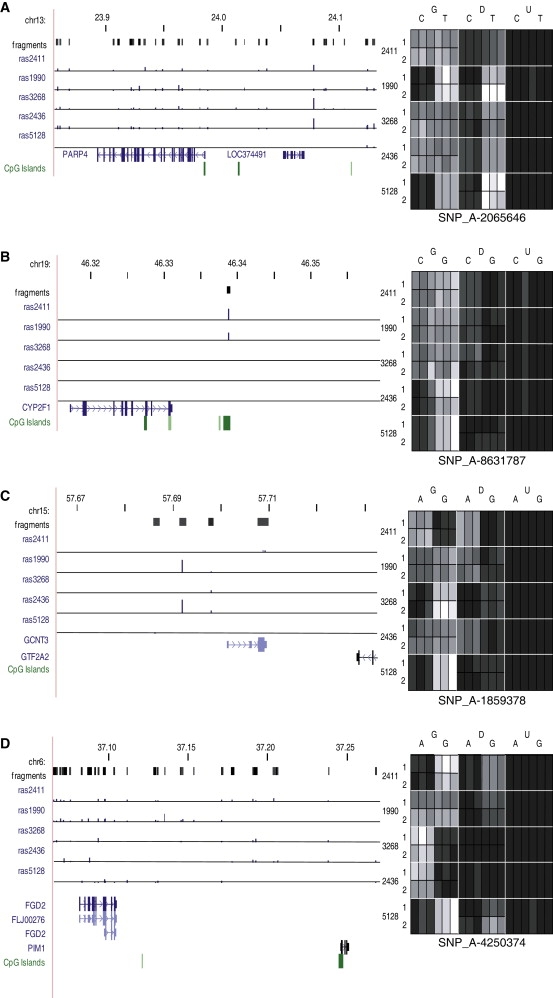
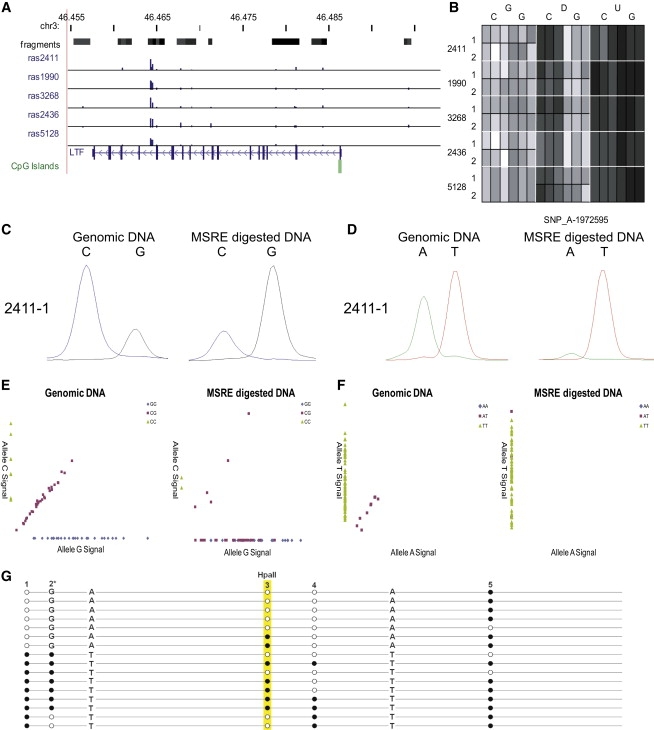
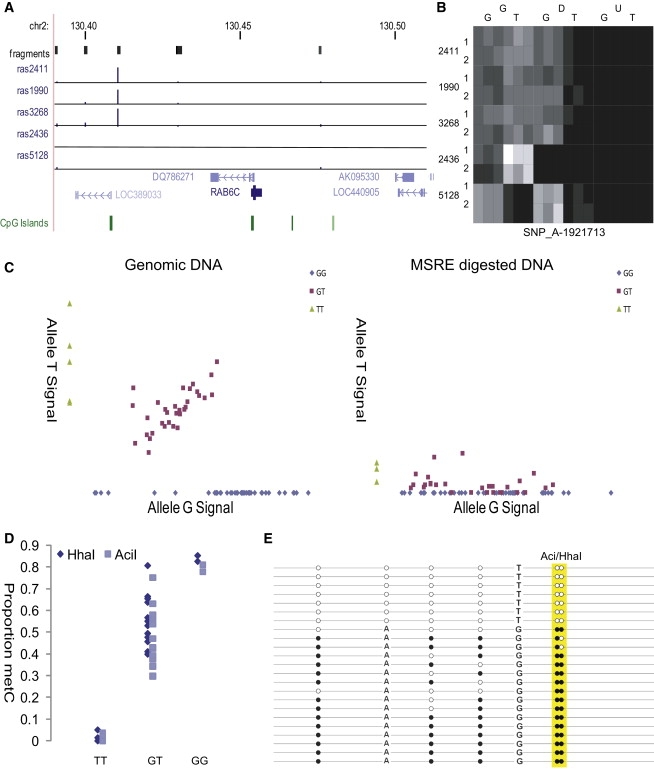
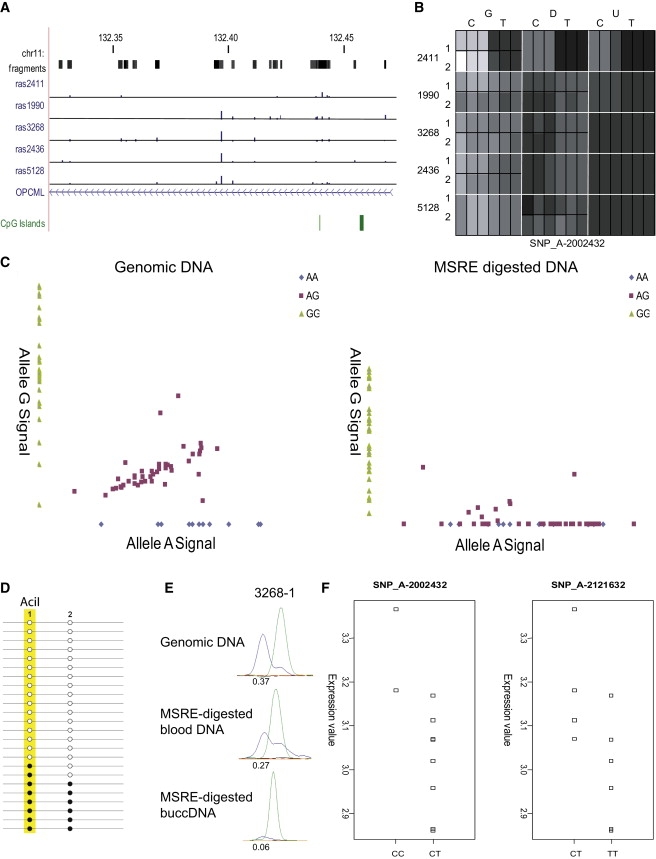
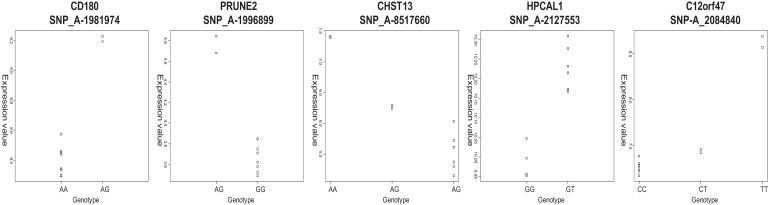
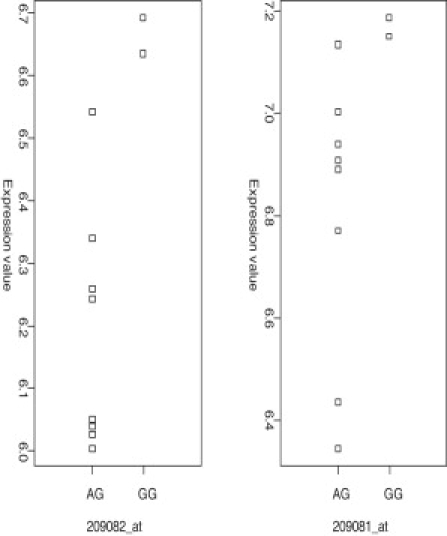
DNA methylation is assumed to be complementary on both alleles across the genome, although there are exceptions, notably in regions subject to genomic imprinting. We present a genome-wide survey of the degree of allelic skewing of DNA methylation with the aim of identifying previously unreported differentially methylated regions (DMRs) associated primarily with genomic imprinting or DNA sequence variation acting in cis. We used SNP microarrays to quantitatively assess allele-specific DNA methylation (ASM) in amplicons covering 7.6% of the human genome following cleavage with a cocktail of methylation-sensitive restriction enzymes (MSREs). Selected findings were verified using bisulfite-mapping and gene-expression analyses, subsequently tested in a second tissue from the same individuals, and replicated in DNA obtained from 30 parent-child trios. Our approach detected clear examples of ASM in the vicinity of known imprinted loci, highlighting the validity of the method. In total, 2,704 (1.5%) of our 183,605 informative and stringently filtered SNPs demonstrate an average relative allele score (RAS) change > or =0.10 following MSRE digestion. In agreement with previous reports, the majority of ASM ( approximately 90%) appears to be cis in nature, and several examples of tissue-specific ASM were identified. Our data show that ASM is a widespread phenomenon, with >35,000 such sites potentially occurring across the genome, and that a spectrum of ASM is likely, with heterogeneity between individuals and across tissues. These findings impact our understanding about the origin of individual phenotypic differences and have implications for genetic studies of complex disease.
Copyright (c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl):245–254. - PubMed
-
- Feinberg A.P. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. - PubMed
-
- Sakatani T., Wei M., Katoh M., Okita C., Wada D., Mitsuya K., Meguro M., Ikeguchi M., Ito H., Tycko B., Oshimura M. Epigenetic heterogeneity at imprinted loci in normal populations. Biochem. Biophys. Res. Commun. 2001;283:1124–1130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
