

Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome
- PMID: 20159255
- PMCID: PMC2878129
- DOI: 10.1016/j.jaci.2009.10.059
Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome
Abstract
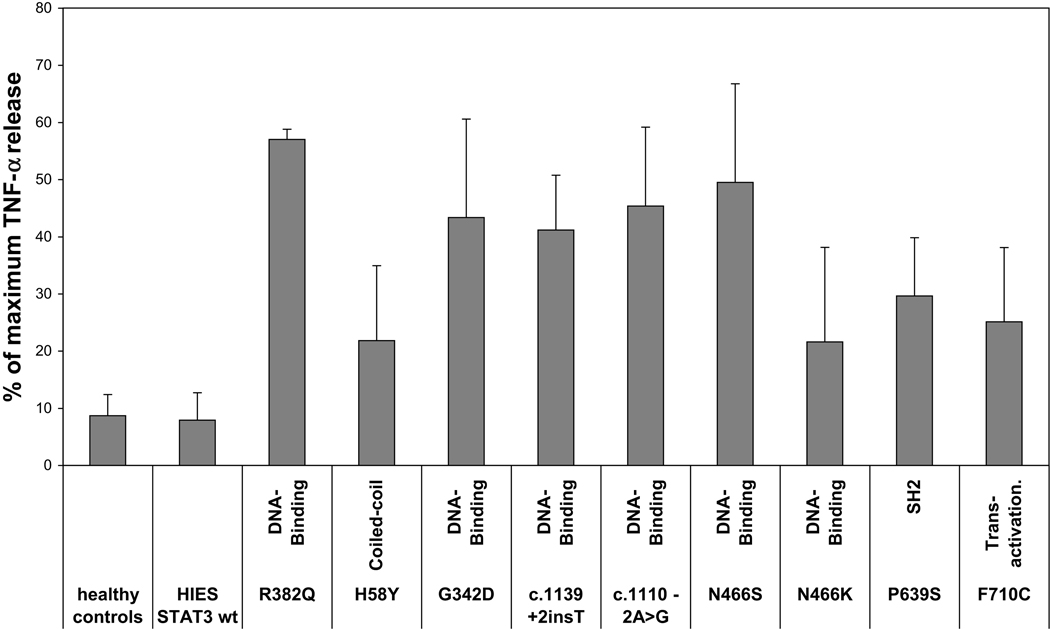
Background: The hyper-IgE syndrome (HIES) is a primary immunodeficiency characterized by infections of the lung and skin, elevated serum IgE, and involvement of the soft and bony tissues. Recently, HIES has been associated with heterozygous dominant-negative mutations in the signal transducer and activator of transcription 3 (STAT3) and severe reductions of T(H)17 cells.
Objective: To determine whether there is a correlation between the genotype and the phenotype of patients with HIES and to establish diagnostic criteria to distinguish between STAT3 mutated and STAT3 wild-type patients.
Methods: We collected clinical data, determined T(H)17 cell numbers, and sequenced STAT3 in 100 patients with a strong clinical suspicion of HIES and serum IgE >1000 IU/mL. We explored diagnostic criteria by using a machine-learning approach to identify which features best predict a STAT3 mutation.
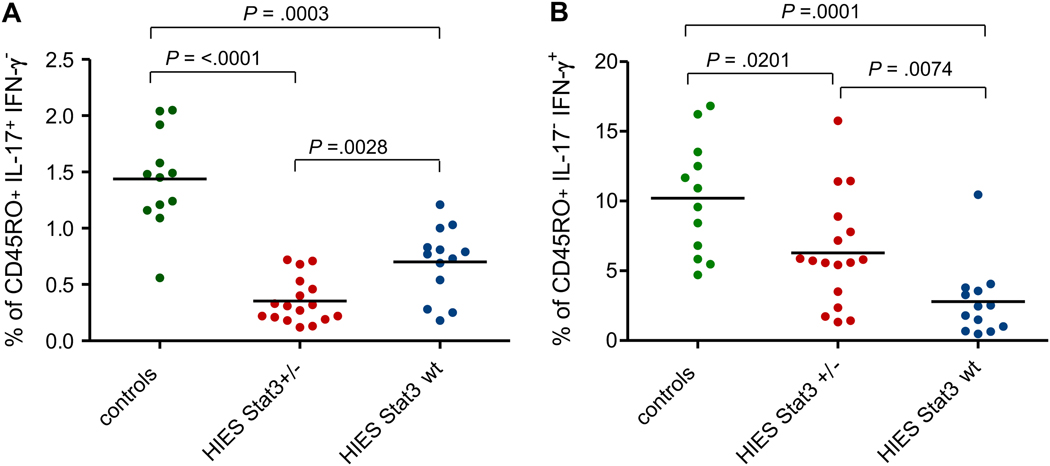
Results: In 64 patients, we identified 31 different STAT3 mutations, 18 of which were novel. These included mutations at splice sites and outside the previously implicated DNA-binding and Src homology 2 domains. A combination of 5 clinical features predicted STAT3 mutations with 85% accuracy. T(H)17 cells were profoundly reduced in patients harboring STAT3 mutations, whereas 10 of 13 patients without mutations had low (<1%) T(H)17 cells but were distinct by markedly reduced IFN-gamma-producing CD4(+)T cells.
Conclusion: We propose the following diagnostic guidelines for STAT3-deficient HIES. Possible: IgE >1000IU/mL plus a weighted score of clinical features >30 based on recurrent pneumonia, newborn rash, pathologic bone fractures, characteristic face, and high palate. Probable: These characteristics plus lack of T(H)17 cells or a family history for definitive HIES. Definitive: These characteristics plus a dominant-negative heterozygous mutation in STAT3.
Copyright 2010 American Academy of Allergy, Asthma & Immunology. Published by Mosby, Inc. All rights reserved.
Conflict of interest statement
All authors declare that they have no conflict of interest.
Figures

References
-
- Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections – an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. - PubMed
-
- Davis SD, Schaller J, Wedgwood RJ. Job's syndrome: recurrent, "cold", staphylococcal abscesses. Lancet. 1966;287:1013–1015. - PubMed
-
- Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49:59–70. - PubMed
-
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
