

Inhibitors of Leishmania GDP-mannose pyrophosphorylase identified by high-throughput screening of small-molecule chemical library
- PMID: 20160053
- PMCID: PMC2863604
- DOI: 10.1128/AAC.01634-09
Inhibitors of Leishmania GDP-mannose pyrophosphorylase identified by high-throughput screening of small-molecule chemical library
Abstract
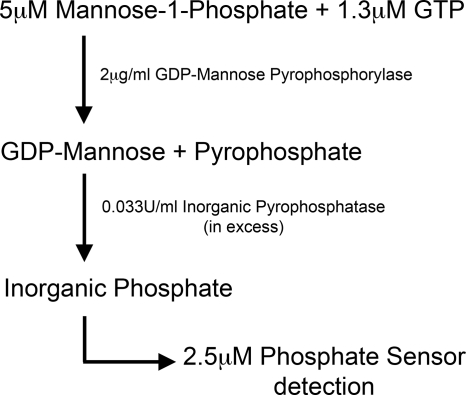
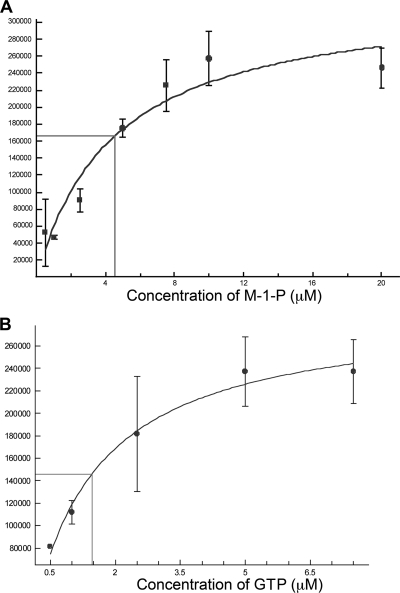
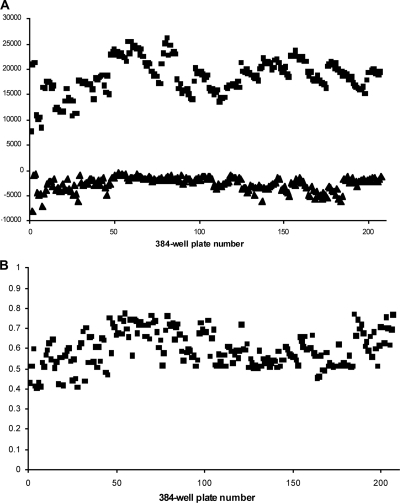
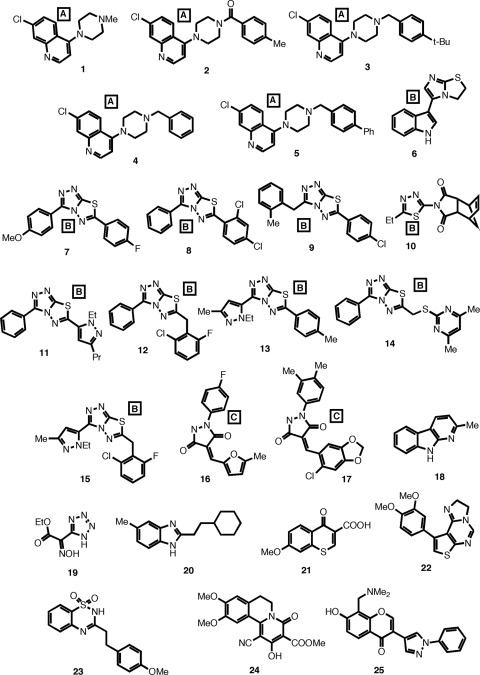
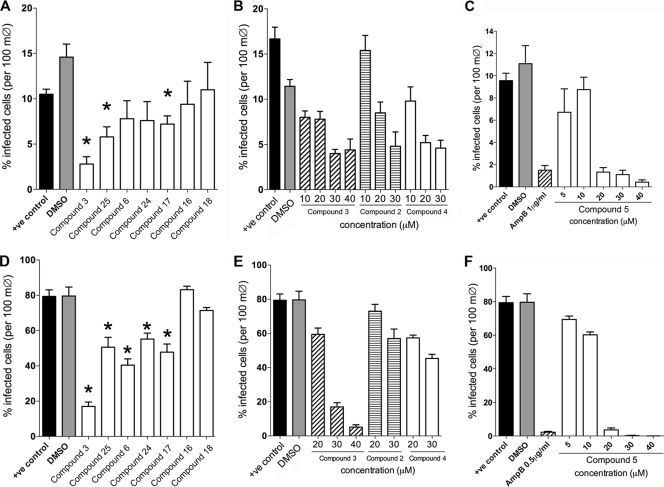
The current treatment for leishmaniasis is based on chemotherapy, which relies on a handful of drugs with serious limitations, such as high cost, toxicity, and a lack of efficacy in regions of endemicity. Therefore, the development of new, effective, and affordable antileishmanial drugs is a global health priority. Leishmania synthesizes a range of mannose-rich glycoconjugates that are essential for parasite virulence and survival. A prerequisite for glycoconjugate biosynthesis is the conversion of monosaccharides to the activated mannose donor, GDP-mannose, the product of a reaction catalyzed by GDP-mannose pyrophosphorylase (GDP-MP). The deletion of the gene encoding GDP-MP in Leishmania led to a total loss of virulence, indicating that the enzyme is an ideal drug target. We developed a phosphate sensor-based high-throughput screening assay to quantify the activity of GDP-MP and screened a library containing approximately 80,000 lead-like compounds for GDP-MP inhibitors. On the basis of their GDP-MP inhibitory properties and chemical structures, the activities of 20 compounds which were not toxic to mammalian cells were tested against ex vivo amastigotes and in macrophage amastigote assays. The most potent compound identified in the primary screen (compound 3), a quinoline derivative, demonstrated dose-dependent activity in both assays (50% inhibitory concentration = 21.9 microM in the macrophage assay) and was shown to be nontoxic to human fibroblasts. In order to elucidate signs of an early structure-activity relationship (SAR) for this class of compounds, we obtained and tested analogues of compound 3 and undertook limited medicinal chemistry optimization, which included the use of a number of SAR probes of the piperazinyl aryl substituent of compound 3. We have identified novel candidate compounds for the design and synthesis of antileishmanial therapeutics.
Figures
Similar articles
-
GDP-Mannose Pyrophosphorylase: A Biologically Validated Target for Drug Development Against Leishmaniasis.Front Cell Infect Microbiol. 2019 May 31;9:186. doi: 10.3389/fcimb.2019.00186. eCollection 2019. Front Cell Infect Microbiol. 2019. PMID: 31214516 Free PMC article. Review.
-
Alkyl-Resorcinol Derivatives as Inhibitors of GDP-Mannose Pyrophosphorylase with Antileishmanial Activities.Molecules. 2021 Mar 11;26(6):1551. doi: 10.3390/molecules26061551. Molecules. 2021. PMID: 33799883 Free PMC article.
-
Properties of GDP-mannose pyrophosphorylase, a critical enzyme and drug target in Leishmania mexicana.J Biol Chem. 2004 Mar 26;279(13):12462-8. doi: 10.1074/jbc.M312365200. Epub 2004 Jan 12. J Biol Chem. 2004. PMID: 14718535
-
Biochemical analysis of leishmanial and human GDP-Mannose Pyrophosphorylases and selection of inhibitors as new leads.Sci Rep. 2017 Apr 7;7(1):751. doi: 10.1038/s41598-017-00848-8. Sci Rep. 2017. PMID: 28389670 Free PMC article.
-
Miltefosine--discovery of the antileishmanial activity of phospholipid derivatives.Trans R Soc Trop Med Hyg. 2006 Dec;100 Suppl 1:S4-8. doi: 10.1016/j.trstmh.2006.03.009. Epub 2006 Aug 14. Trans R Soc Trop Med Hyg. 2006. PMID: 16904717 Review.
Cited by
-
Drug Discovery for Cutaneous Leishmaniasis: A Review of Developments in the Past 15 Years.Microorganisms. 2023 Nov 23;11(12):2845. doi: 10.3390/microorganisms11122845. Microorganisms. 2023. PMID: 38137989 Free PMC article. Review.
-
Identification of Leishmania major UDP-Sugar Pyrophosphorylase Inhibitors Using Biosensor-Based Small Molecule Fragment Library Screening.Molecules. 2019 Mar 12;24(5):996. doi: 10.3390/molecules24050996. Molecules. 2019. PMID: 30871023 Free PMC article.
-
Phosphoglucomutase is absent in Trypanosoma brucei and redundantly substituted by phosphomannomutase and phospho-N-acetylglucosamine mutase.Mol Microbiol. 2012 Aug;85(3):513-34. doi: 10.1111/j.1365-2958.2012.08124.x. Epub 2012 Jul 12. Mol Microbiol. 2012. PMID: 22676716 Free PMC article.
-
In silico analysis of a therapeutic target in Leishmania infantum: the guanosine-diphospho-D-mannose pyrophosphorylase.Parasite. 2012 Feb;19(1):63-70. doi: 10.1051/parasite/2012191063. Parasite. 2012. PMID: 22314241 Free PMC article.
-
Trypanosoma cruzi Phosphomannomutase and Guanosine Diphosphate-Mannose Pyrophosphorylase Ligandability Assessment.Antimicrob Agents Chemother. 2019 Sep 23;63(10):e01082-19. doi: 10.1128/AAC.01082-19. Print 2019 Oct. Antimicrob Agents Chemother. 2019. PMID: 31405854 Free PMC article.
References
-
- Reference deleted.
-
- Armutlu, P., M. E. Ozdemir, S. Ozdas, I. H. Kavakli, and M. Turkay. 2009. Discovery of novel CYP17 inhibitors for the treatment of prostate cancer with structure-based design. Lett. Drug Des. Discov. 6:337-344.
-
- Bertucci, A., A. Innocenti, D. Zoccola, A. Scozzafava, S. Tambutte, and C. T. Supuran. 2009. Carbonic anhydrase inhibitors. Inhibition studies of a coral secretory isoform by sulfonamides. Bioorg. Med. Chem. 17:5054-5058. - PubMed
-
- Cheeseright, T. J., M. Holm, F. Lehmann, S. Luik, M. Gottert, J. L. Melville, and S. Laufer. 2009. Novel lead structures for p38 MAP kinase via FieldScreen virtual screening. J. Med. Chem. 52:4200-4209. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
