

TGF-beta regulates T-cell neurokinin-1 receptor internalization and function
- PMID: 20160079
- PMCID: PMC2840170
- DOI: 10.1073/pnas.0905877107
TGF-beta regulates T-cell neurokinin-1 receptor internalization and function
Abstract
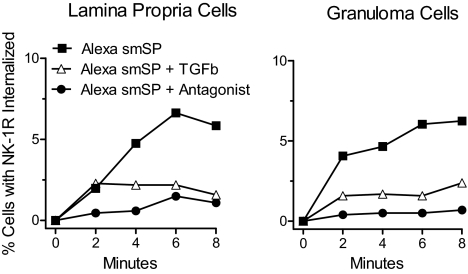
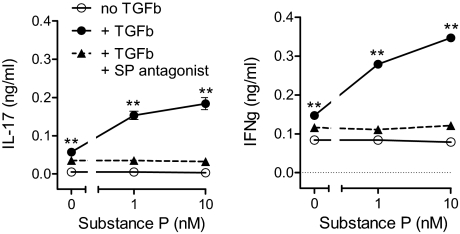
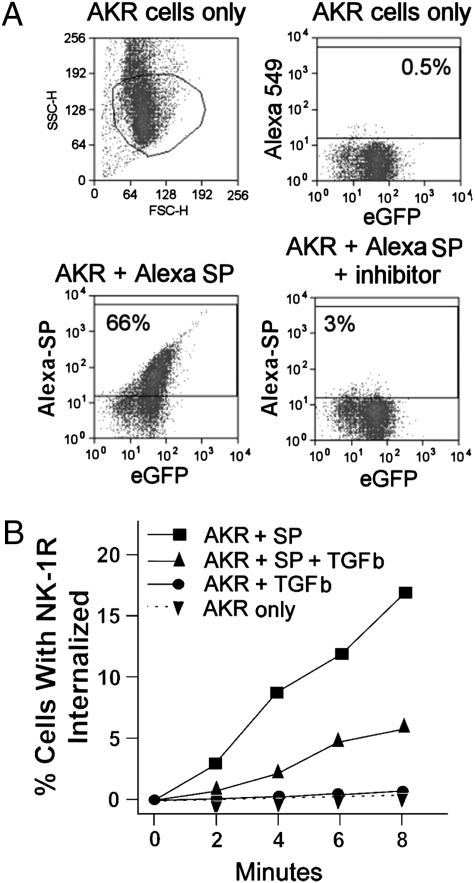
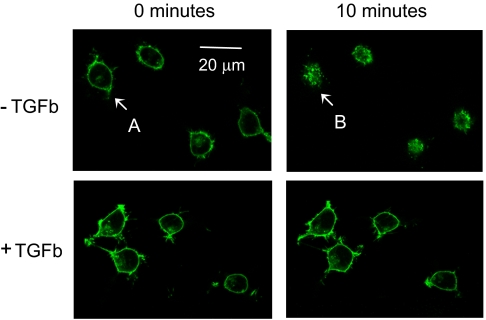
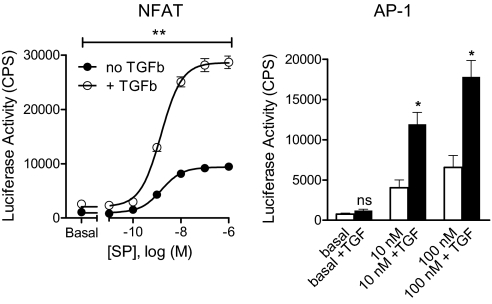
Substance P (SP) is a proinflammatory mediator implicated in inflammatory bowel disease (IBD) and other inflammatory states. SP acts by stimulating the neurokinin-1 receptor (NK-1R) on T lymphocytes and other cell types, and regulates these cells in a complex interplay with multiple cytokines. The mechanisms of interaction among these inflammatory mediators are not yet fully understood. Here, we demonstrate that function of the NK-1R, a member of the G protein-coupled receptor (GPCR) superfamily, is modulated by TGF-beta. The latter acts not on a GPCR but via serine-threonine kinase-class receptors. By flow confocal image analysis, we demonstrate that TGF-beta delays SP-induced NK-1R internalization on mucosal T cells isolated from a mouse model of IBD and on granuloma T cells in murine schistosomiasis. Furthermore, luciferase reporter-gene assays revealed that NK-1R stimulation activates the nuclear factor of activated T cell- and activator protein-1-dependent signaling pathways, which are known triggers of effector T-cell cytokine production. TGF-beta markedly increases SP-induced activation of these signaling cascades, suggesting that delayed NK-1R internalization results in enhanced signaling. Providing a link to amplified immune function, SP and TGF-beta, when applied in combination, trigger a strong release of the proinflammatory cytokines IFN-gamma and IL17 from intestinal inflammatory T cells, whereas either agonist alone shows no effect. These observations establish precedent that members of two distinct receptor superfamilies can interact via a previously unrecognized mechanism, and reveal a paradigm of GPCR transregulation that is relevant to IBD and possibly other disease processes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- O'Connor TM, et al. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201:167–180. - PubMed
-
- Almeida TA, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Curr Med Chem. 2004;11:2045–2081. - PubMed
-
- Smith VC, Sagot MA, Couraud JY, Buchan AM. Localization of the neurokinin 1 (NK-1) receptor in the human antrum and duodenum. Neurosci Lett. 1998;253:49–52. - PubMed
-
- Goode T, et al. Substance P (neurokinin-1) receptor is a marker of human mucosal but not peripheral mononuclear cells: molecular quantitation and localization. J Immunol. 1998;161:2232–2240. - PubMed
-
- Cook GA, et al. Molecular evidence that granuloma T lymphocytes in murine schistosomiasis mansoni express an authentic substance P (NK-1) receptor. J Immunol. 1994;152:1830–1835. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
