

Autophagy in the lung
- PMID: 20160144
- PMCID: PMC3137145
- DOI: 10.1513/pats.200909-101JS
Autophagy in the lung
Abstract
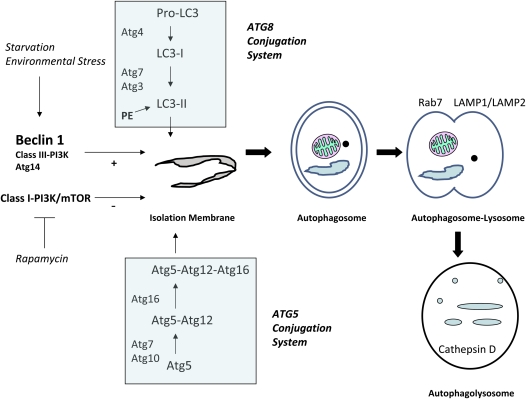
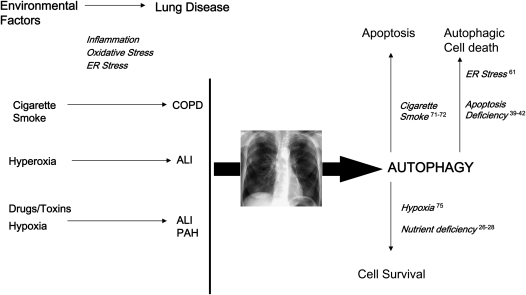
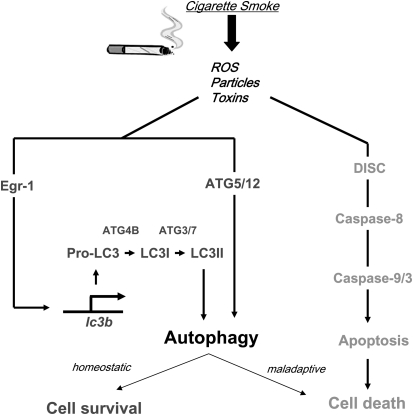
Autophagy is a cellular process for the disposal of damaged organelles or denatured proteins through a lysosomal degradation pathway. By reducing endogenous macromolecules to their basic components (i.e., amino acids, lipids), autophagy serves a homeostatic function by ensuring cell survival during starvation. Increased autophagy can be found in dying cells, although the relationships between autophagy and programmed cell death remain unclear. To date, few studies have examined the regulation and functional significance of autophagy in human lung disease. The lung, a complex organ that functions primarily in gas exchange, consists of diverse cell types (i.e., endothelial, epithelial, mesenchymal, inflammatory). In lung cells, autophagy may represent a general inducible adaptive response to injury resulting from exposure to stress agents, including hypoxia, oxidants, inflammation, ischemia-reperfusion, endoplasmic reticulum stress, pharmaceuticals, or inhaled xenobiotics (i.e., air pollution, cigarette smoke). In recent studies, we have observed increased autophagy in mouse lungs subjected to chronic cigarette smoke exposure, and in pulmonary epithelial cells exposed to cigarette smoke extract. Knockdown of autophagic proteins inhibited apoptosis in response to cigarette smoke exposure in vitro, suggesting that increased autophagy was associated with epithelial cell death. We have also observed increased morphological and biochemical markers of autophagy in human lung specimens from patients with chronic obstructive pulmonary disease (COPD). We hypothesize that increased autophagy contributes to COPD pathogenesis by promoting epithelial cell death. Further research will examine whether autophagy plays a homeostatic or maladaptive role in COPD and other human lung diseases.
Figures
Similar articles
-
Autophagy in chronic obstructive pulmonary disease: homeostatic or pathogenic mechanism?Autophagy. 2009 Feb;5(2):235-7. doi: 10.4161/auto.5.2.7495. Epub 2009 Feb 26. Autophagy. 2009. PMID: 19066468
-
Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease.PLoS One. 2008 Oct 2;3(10):e3316. doi: 10.1371/journal.pone.0003316. PLoS One. 2008. PMID: 18830406 Free PMC article.
-
Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease.Expert Rev Respir Med. 2010 Oct;4(5):573-84. doi: 10.1586/ers.10.61. Expert Rev Respir Med. 2010. PMID: 20923337 Free PMC article. Review.
-
Autophagy in vascular disease.Proc Am Thorac Soc. 2010 Feb;7(1):40-7. doi: 10.1513/pats.200909-100JS. Proc Am Thorac Soc. 2010. PMID: 20160147 Free PMC article. Review.
-
Deadly triplex: smoke, autophagy and apoptosis.Autophagy. 2011 Apr;7(4):436-7. doi: 10.4161/auto.7.4.14501. Epub 2011 Apr 1. Autophagy. 2011. PMID: 21200154
Cited by
-
The interplay between oxidative stress and autophagy in chronic obstructive pulmonary disease.Front Physiol. 2022 Sep 26;13:1004275. doi: 10.3389/fphys.2022.1004275. eCollection 2022. Front Physiol. 2022. PMID: 36225291 Free PMC article.
-
Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice.Am J Physiol Lung Cell Mol Physiol. 2014 Sep 1;307(5):L355-63. doi: 10.1152/ajplung.00046.2014. Epub 2014 Jul 3. Am J Physiol Lung Cell Mol Physiol. 2014. PMID: 24993132 Free PMC article.
-
Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma.Korean J Intern Med. 2016 Mar;31(2):375-85. doi: 10.3904/kjim.2014.390. Epub 2015 Dec 23. Korean J Intern Med. 2016. PMID: 26701229 Free PMC article.
-
Cross talk between autophagy and apoptosis in pulmonary hypertension.Pulm Circ. 2012 Oct;2(4):407-14. doi: 10.4103/2045-8932.105029. Pulm Circ. 2012. PMID: 23372925 Free PMC article.
-
Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis.Autophagy. 2015 Apr 3;11(4):670-84. doi: 10.1080/15548627.2015.1034409. Autophagy. 2015. PMID: 25906080 Free PMC article.
References
-
- Menzel DB, Amdur MO. Toxic response of the respiratory system. In: Klaassen K, Amdur MO, Doull J, editors. Casarett and Doull's toxicology, the basic science of poisons, 3rd ed. New York, NY, MacMillan Publishing Company; 1986. pp. 330–358.
-
- Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. Cell number and cell characteristics of the normal human lung. Am Rev Respir Dis 1982;126:332–337. - PubMed
-
- Pagano A, Barazzone-Argiroffo C. Alveolar cell death in hyperoxia-induced lung injury. Ann N Y Acad Sci 2003;1010:405–416. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical