

Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles
- PMID: 20161451
- PMCID: PMC2790287
- DOI: 10.1021/ct9001812
Quantifying Correlations Between Allosteric Sites in Thermodynamic Ensembles
Abstract
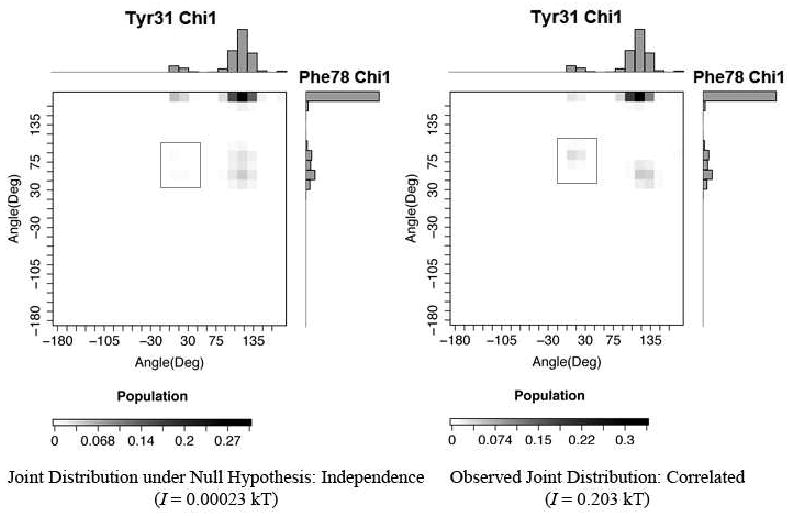
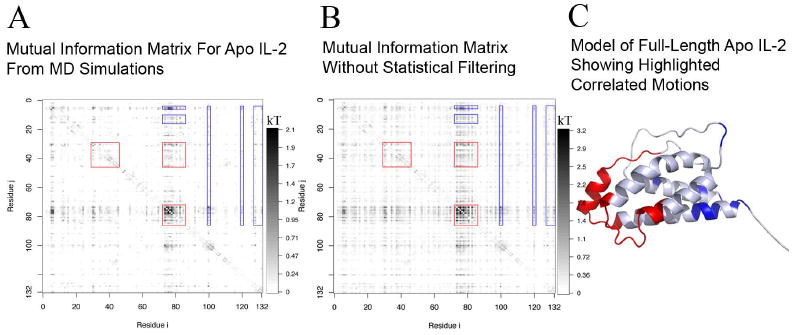
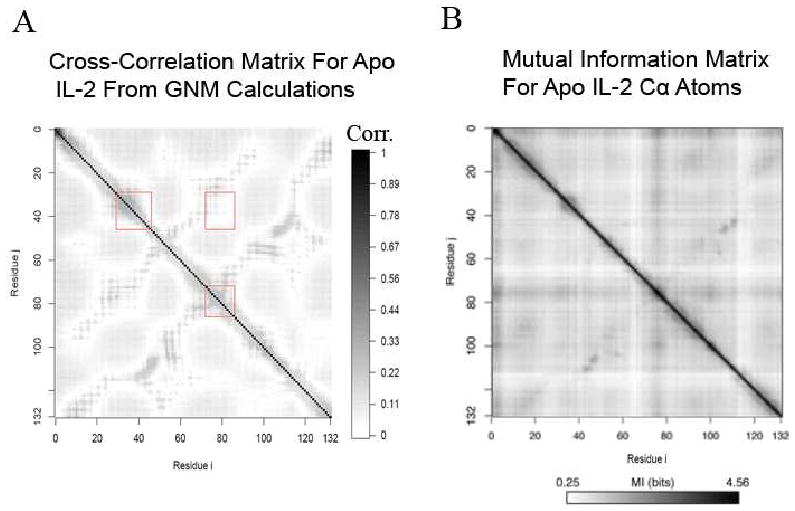
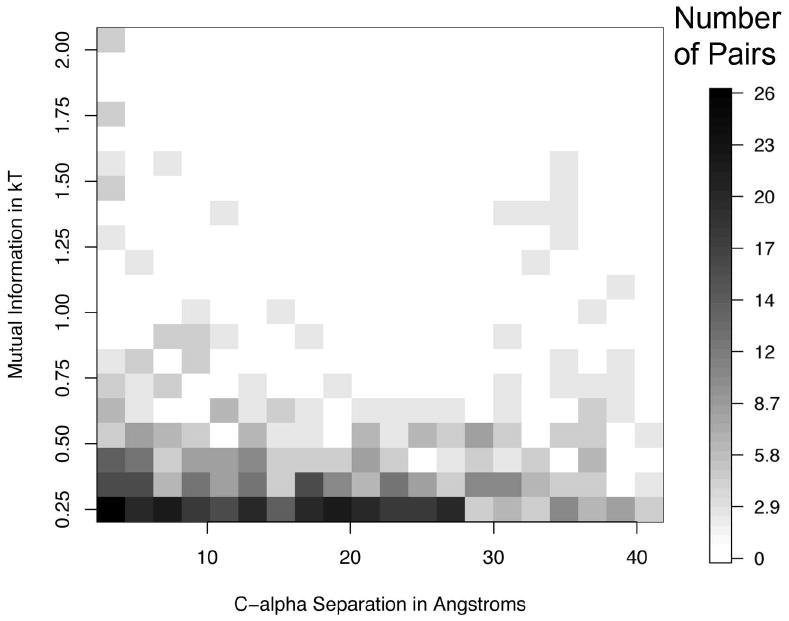
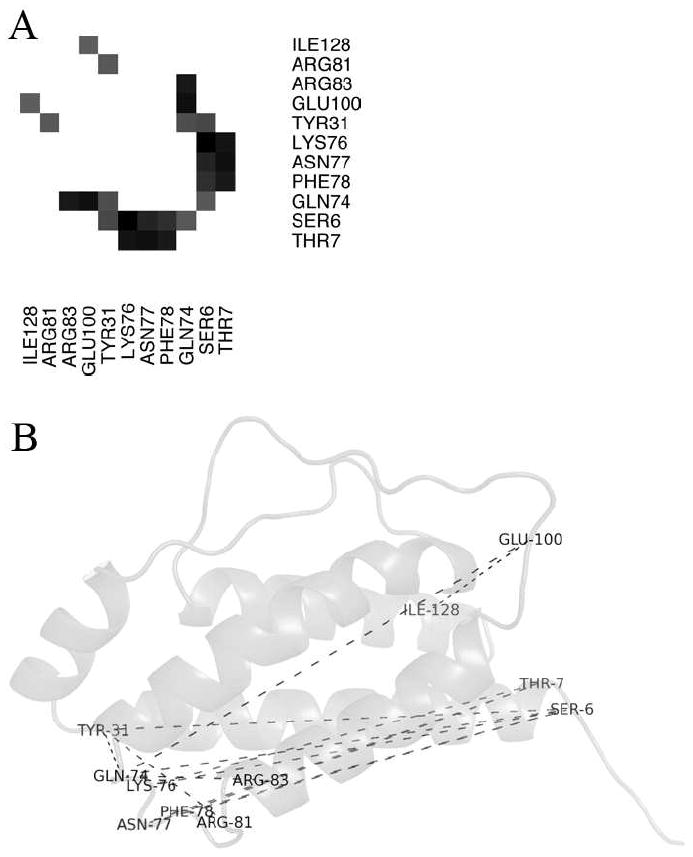
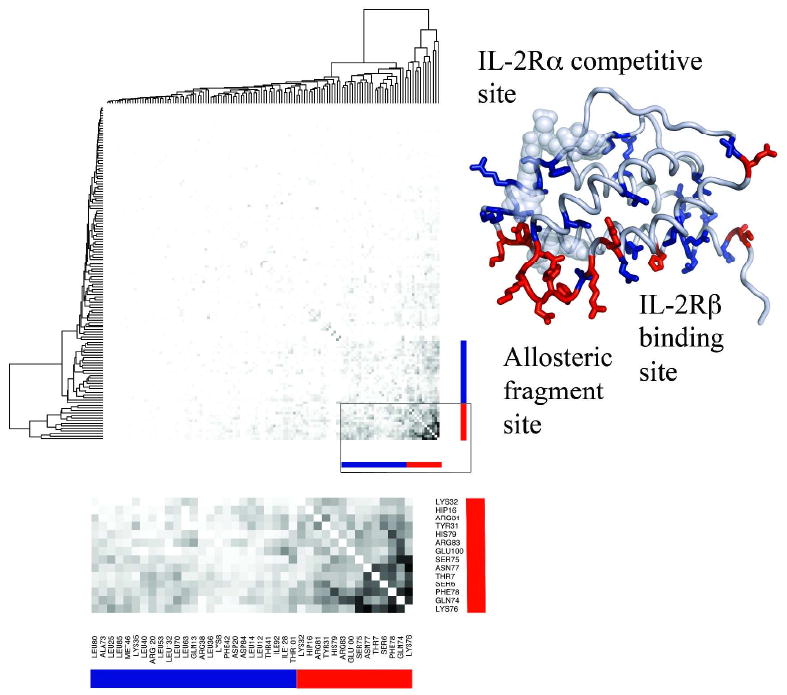
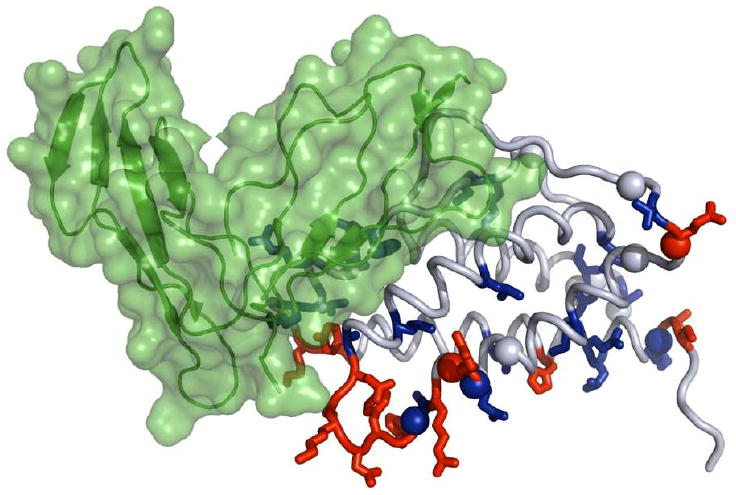
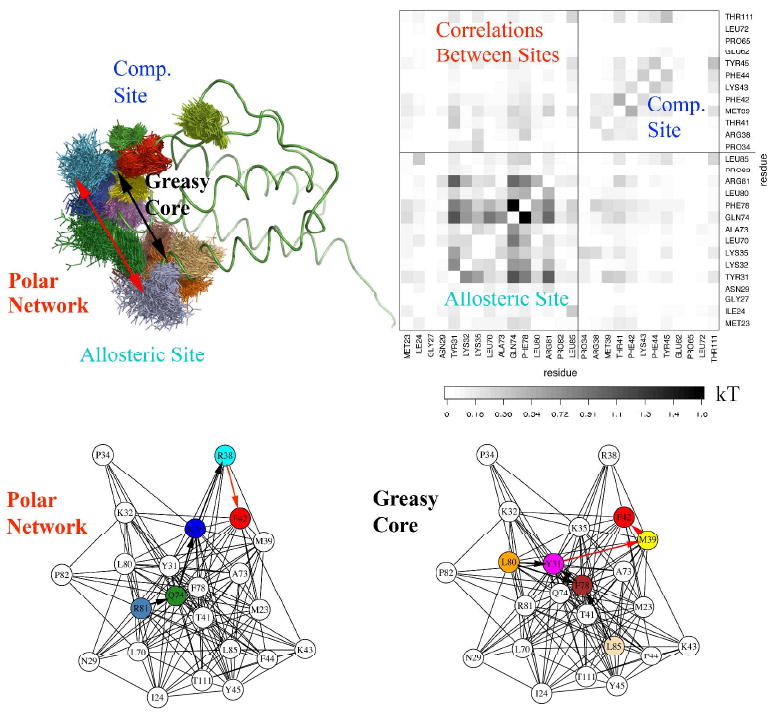
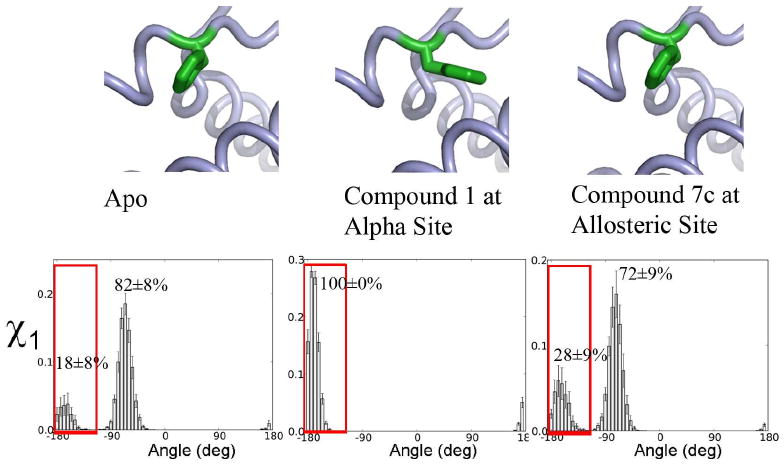
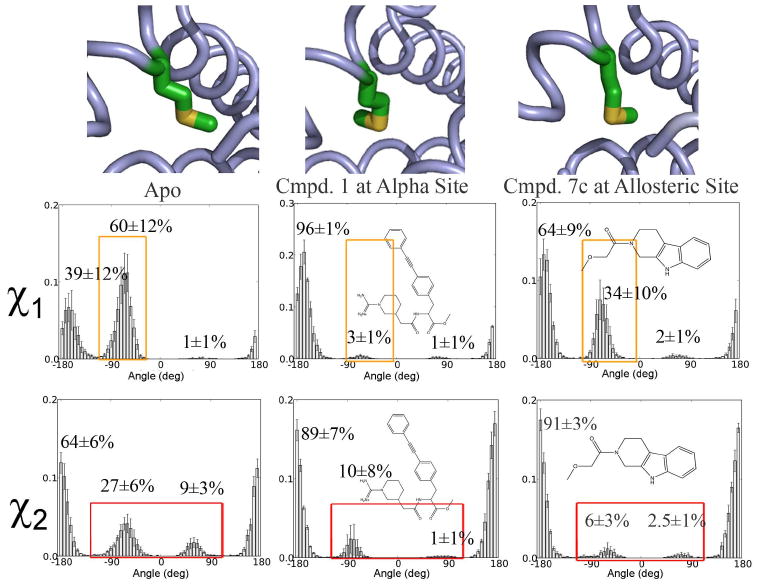
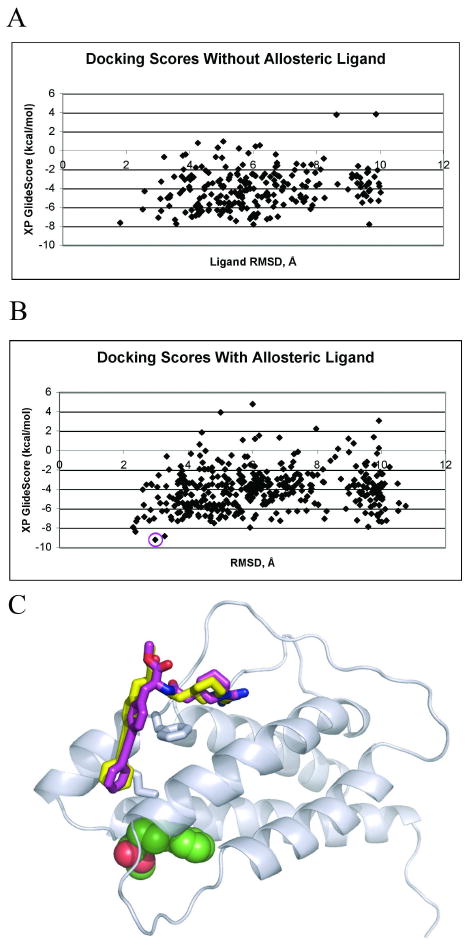
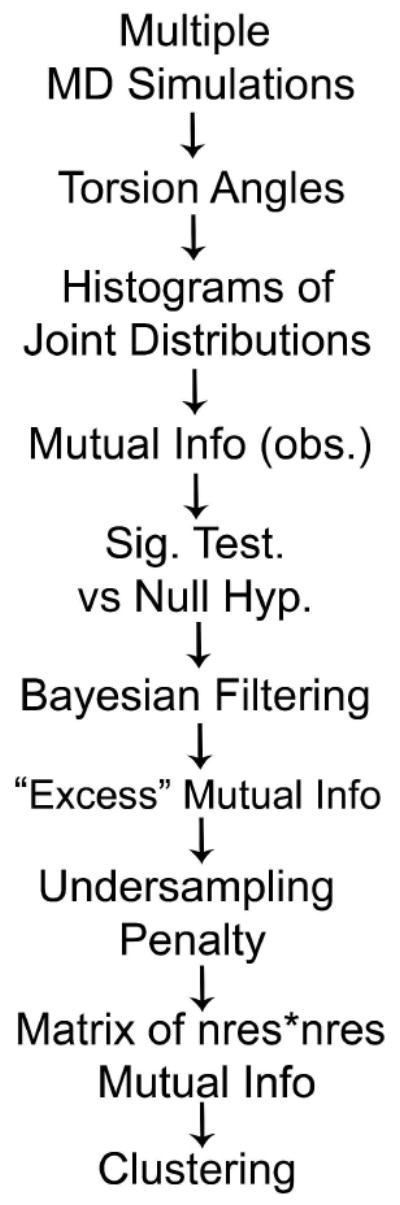
Allostery describes altered protein function at one site due to a perturbation at another site. One mechanism of allostery involves correlated motions, which can occur even in the absence of substantial conformational change. We present a novel method, "MutInf", to identify statistically significant correlated motions from equilibrium molecular dynamics simulations. Our approach analyzes both backbone and sidechain motions using internal coordinates to account for the gear-like twists that can take place even in the absence of the large conformational changes typical of traditional allosteric proteins. We quantify correlated motions using a mutual information metric, which we extend to incorporate data from multiple short simulations and to filter out correlations that are not statistically significant. Applying our approach to uncover mechanisms of cooperative small molecule binding in human interleukin-2, we identify clusters of correlated residues from 50 ns of molecular dynamics simulations. Interestingly, two of the clusters with the strongest correlations highlight known cooperative small-molecule binding sites and show substantial correlations between these sites. These cooperative binding sites on interleukin-2 are correlated not only through the hydrophobic core of the protein but also through a dynamic polar network of hydrogen bonding and electrostatic interactions. Since this approach identifies correlated conformations in an unbiased, statistically robust manner, it should be a useful tool for finding novel or "orphan" allosteric sites in proteins of biological and therapeutic importance.
Figures
Similar articles
-
Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated.J Chem Inf Model. 2016 Sep 26;56(9):1725-33. doi: 10.1021/acs.jcim.6b00039. Epub 2016 Sep 12. J Chem Inf Model. 2016. PMID: 27580047
-
Modulation of global low-frequency motions underlies allosteric regulation: demonstration in CRP/FNR family transcription factors.PLoS Biol. 2013 Sep;11(9):e1001651. doi: 10.1371/journal.pbio.1001651. Epub 2013 Sep 10. PLoS Biol. 2013. PMID: 24058293 Free PMC article.
-
Exploring Conformational Landscapes and Cryptic Binding Pockets in Distinct Functional States of the SARS-CoV-2 Omicron BA.1 and BA.2 Trimers: Mutation-Induced Modulation of Protein Dynamics and Network-Guided Prediction of Variant-Specific Allosteric Binding Sites.Viruses. 2023 Sep 27;15(10):2009. doi: 10.3390/v15102009. Viruses. 2023. PMID: 37896786 Free PMC article.
-
Detecting Functional Dynamics in Proteins with Comparative Perturbed-Ensembles Analysis.Acc Chem Res. 2019 Dec 17;52(12):3455-3464. doi: 10.1021/acs.accounts.9b00485. Epub 2019 Dec 3. Acc Chem Res. 2019. PMID: 31793290 Review.
-
Folding funnels and conformational transitions via hinge-bending motions.Cell Biochem Biophys. 1999;31(2):141-64. doi: 10.1007/BF02738169. Cell Biochem Biophys. 1999. PMID: 10593256 Review.
Cited by
-
Dynamic allostery-based molecular workings of kinase:peptide complexes.Proc Natl Acad Sci U S A. 2019 Jul 23;116(30):15052-15061. doi: 10.1073/pnas.1900163116. Epub 2019 Jul 8. Proc Natl Acad Sci U S A. 2019. PMID: 31285328 Free PMC article.
-
Computational and Experimental Characterization of Patient Derived Mutations Reveal an Unusual Mode of Regulatory Spine Assembly and Drug Sensitivity in EGFR Kinase.Biochemistry. 2017 Jan 10;56(1):22-32. doi: 10.1021/acs.biochem.6b00572. Epub 2016 Dec 22. Biochemistry. 2017. PMID: 27936599 Free PMC article.
-
Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics.PLoS Comput Biol. 2016 Apr 28;12(4):e1004619. doi: 10.1371/journal.pcbi.1004619. eCollection 2016 Apr. PLoS Comput Biol. 2016. PMID: 27124275 Free PMC article. Review.
-
Impact of mutations on the allosteric conformational equilibrium.J Mol Biol. 2013 Feb 8;425(3):647-61. doi: 10.1016/j.jmb.2012.11.041. Epub 2012 Dec 7. J Mol Biol. 2013. PMID: 23228330 Free PMC article.
-
Cushing's syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity.Sci Adv. 2019 Aug 28;5(8):eaaw9298. doi: 10.1126/sciadv.aaw9298. eCollection 2019 Aug. Sci Adv. 2019. PMID: 31489371 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous