

Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy
- PMID: 20161659
- PMCID: PMC2818805
- DOI: 10.1126/scitranslmed.3000208
Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy
Abstract
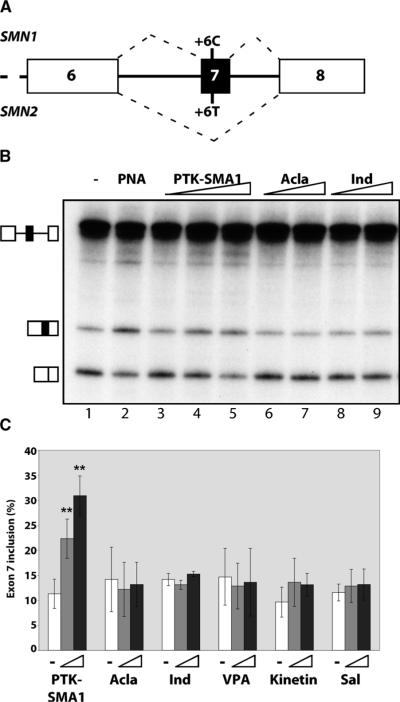
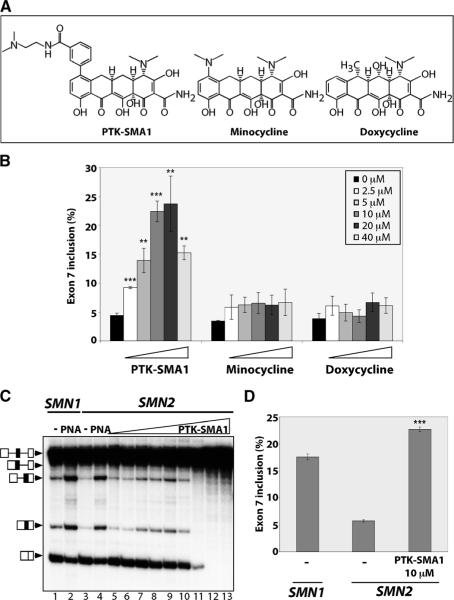
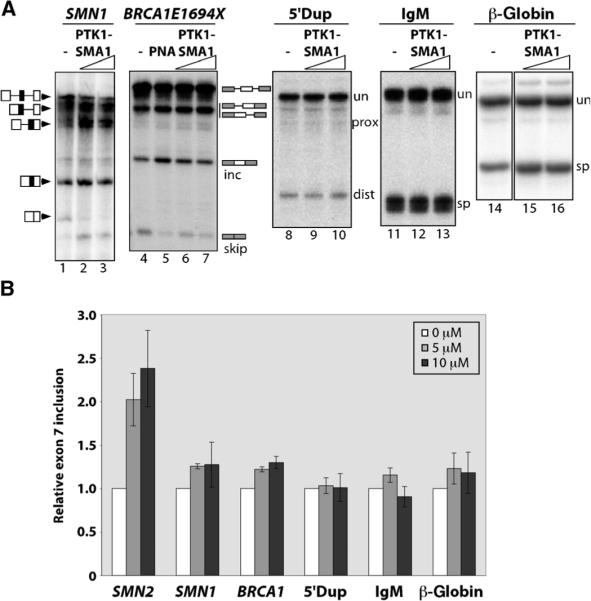
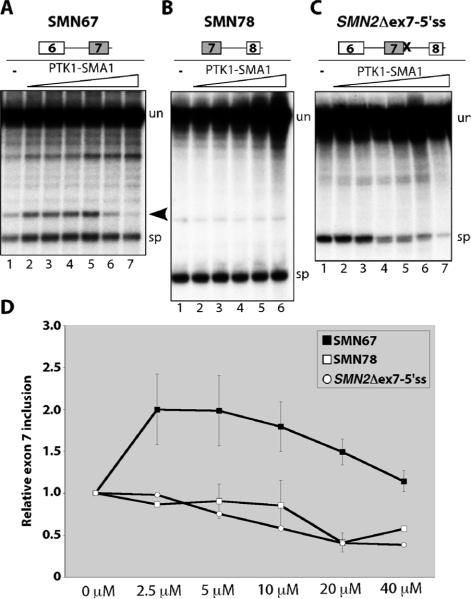
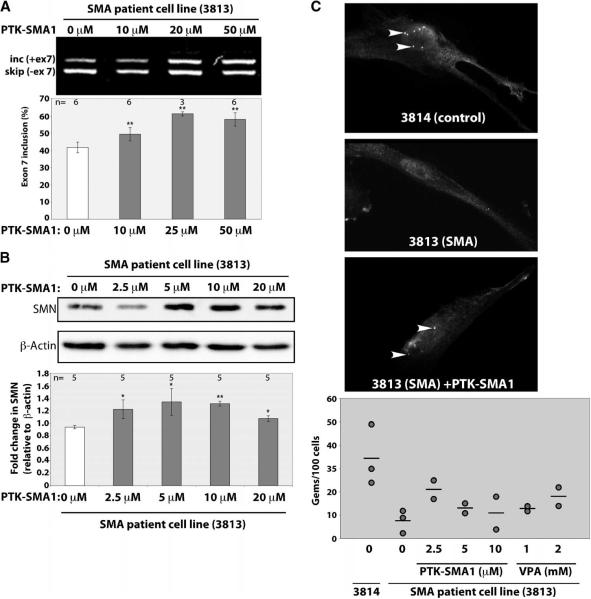
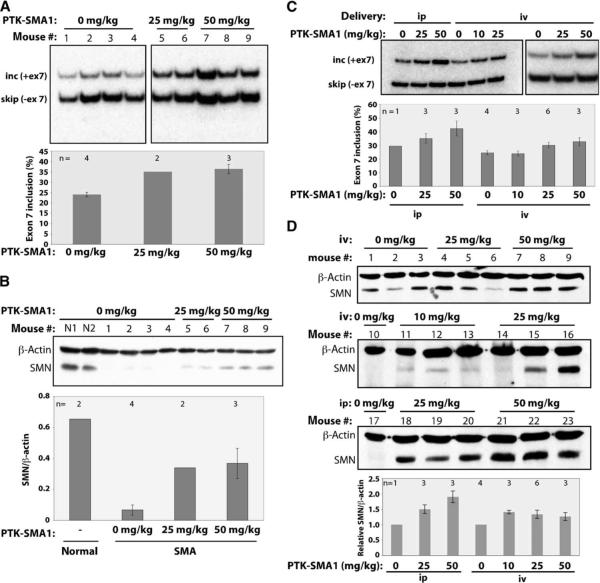
There is at present no cure or effective therapy for spinal muscular atrophy (SMA), a neurodegenerative disease that is the leading genetic cause of infant mortality. SMA usually results from loss of the SMN1 (survival of motor neuron 1) gene, which leads to selective motor neuron degeneration. SMN2 is nearly identical to SMN1 but has a nucleotide replacement that causes exon 7 skipping, resulting in a truncated, unstable version of the SMA protein. SMN2 is present in all SMA patients, and correcting SMN2 splicing is a promising approach for SMA therapy. We identified a tetracycline-like compound, PTK-SMA1, which stimulates exon 7 splicing and increases SMN protein levels in vitro and in vivo in mice. Unlike previously identified molecules that stimulate SMN production via SMN2 promoter activation or undefined mechanisms, PTK-SMA1 is a unique therapeutic candidate in that it acts by directly stimulating splicing of exon 7. Synthetic small-molecule compounds such as PTK-SMA1 offer an alternative to antisense oligonucleotide therapies that are being developed as therapeutics for a number of disease-associated splicing defects.
Figures
References
-
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
-
- Munsat TL, Davies KE. Neuromuscul. Disord; International SMA consortium meeting; Bonn, Germany. 26–28 June 1992; 1992. pp. 423–428. - PubMed
-
- Neuenkirchen N, Chari A, Fischer U. Deciphering the assembly pathway of Sm-class U snRNPs. FEBS Lett. 2008;582:1997–2003. - PubMed
-
- Wahl MC, Will CL, Lührmann R. The spliceosome: Design principles of a dynamic RNP machine. Cell. 2009;136:701–718. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
