

Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing
- PMID: 20161664
- PMCID: PMC2819115
- DOI: 10.1126/scitranslmed.3000540
Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing
Abstract
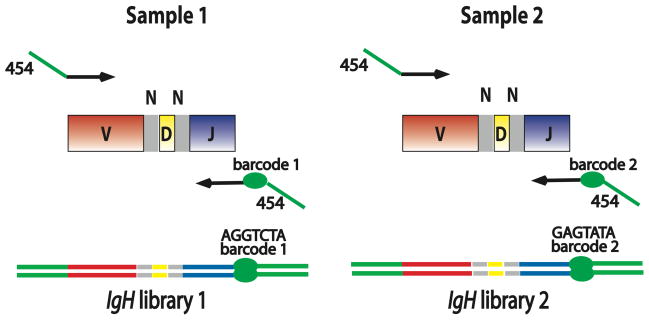
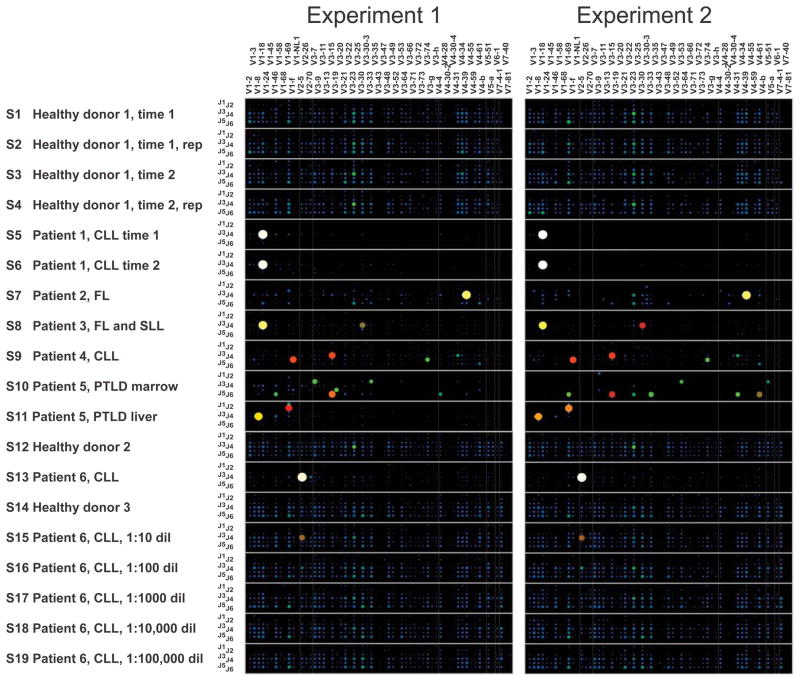
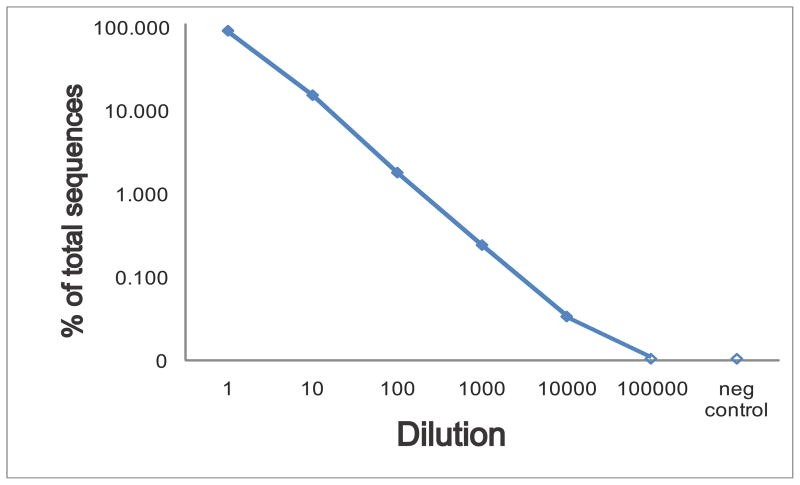
The complex repertoire of immune receptors generated by B and T cells enables recognition of diverse threats to the host organism. In this work, we show that massively parallel DNA sequencing of rearranged immune receptor loci can provide direct detection and tracking of immune diversity and expanded clonal lymphocyte populations in physiological and pathological contexts. DNA was isolated from blood and tissue samples, a series of redundant primers was used to amplify diverse DNA rearrangements, and the resulting mixtures of barcoded amplicons were sequenced using long-read ultra deep sequencing. Individual DNA molecules were then characterized on the basis of DNA segments that had been joined to make a functional (or nonfunctional) immune effector. Current experimental designs can accommodate up to 150 samples in a single sequence run, with the depth of sequencing sufficient to identify stable and dynamic aspects of the immune repertoire in both normal and diseased circumstances. These data provide a high-resolution picture of immune spectra in normal individuals and in patients with hematological malignancies, illuminating, in the latter case, both the initial behavior of clonal tumor populations and the later suppression or re-emergence of such populations after treatment.
Conflict of interest statement
Boyd, Marshall, Merker, Maniar, Zhang, Sahaf, Jones, Nadeau, Nguyen, Miklos, Zehnder, Fire: None Simen, Hanczaruk, and Egholm are employees of 454 Life Sciences, A Roche Company
Figures
References
-
- Schatz DG. Antigen receptor genes and the evolution of a recombinase. Semin Immunol. 2004;16:245–256. - PubMed
-
- Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. - PubMed
-
- Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999;286:958–961. - PubMed
-
- Brezinschek HP, Brezinschek RI, Lipsky PE. Analysis of the heavy chain repertoire of human peripheral B cells using single-cell polymerase chain reaction. J Immunol. 1995;155:190–202. - PubMed
-
- Lim A, Luderschmidt S, Weidinger A, Schnopp C, Ring J, Hein R, Ollert M, Mempel M. The IgE repertoire in PBMCs of atopic patients is characterized by individual rearrangements without variable region of the heavy immunoglobulin chain bias. J Allergy Clin Immunol. 2007;120:696–706. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
