

Combinatorial effects of double cardiomyopathy mutant alleles in rodent myocytes: a predictive cellular model of myofilament dysregulation in disease
- PMID: 20161772
- PMCID: PMC2818843
- DOI: 10.1371/journal.pone.0009140
Combinatorial effects of double cardiomyopathy mutant alleles in rodent myocytes: a predictive cellular model of myofilament dysregulation in disease
Abstract
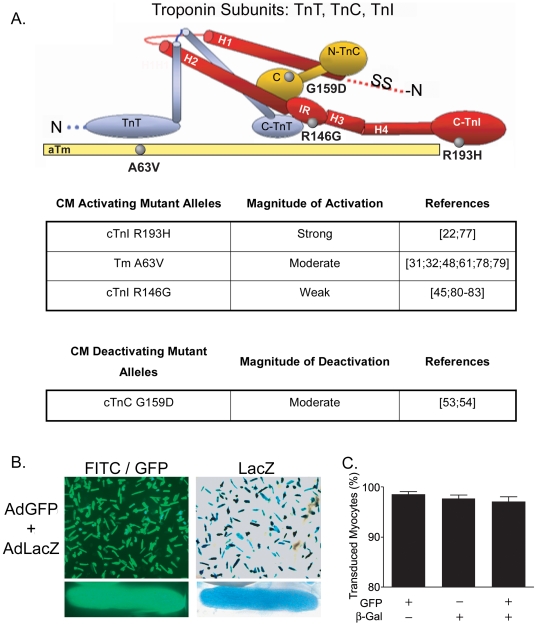
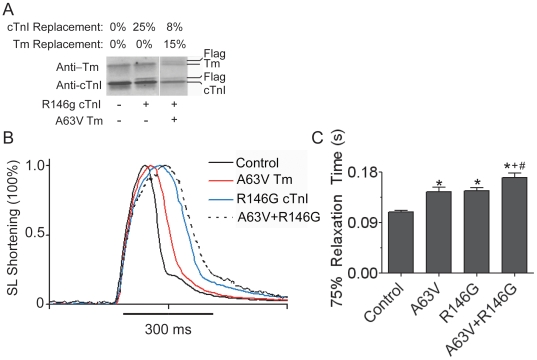
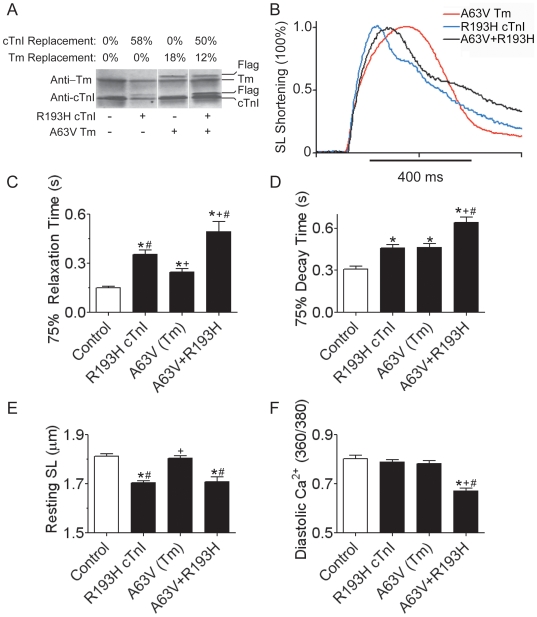
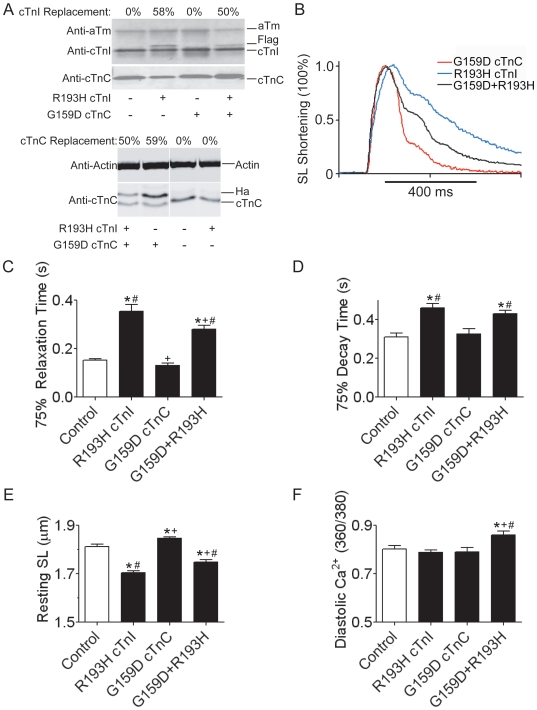
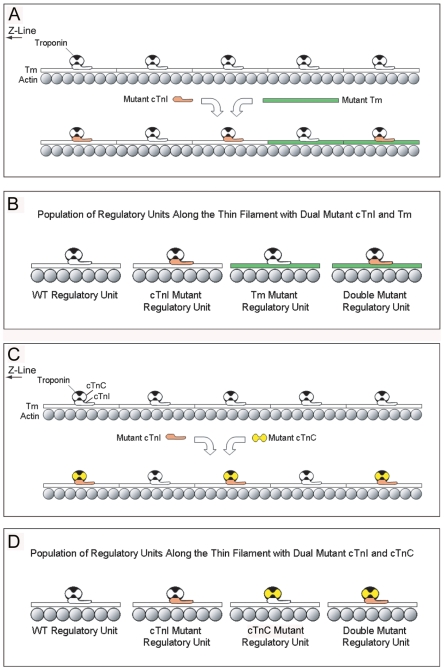
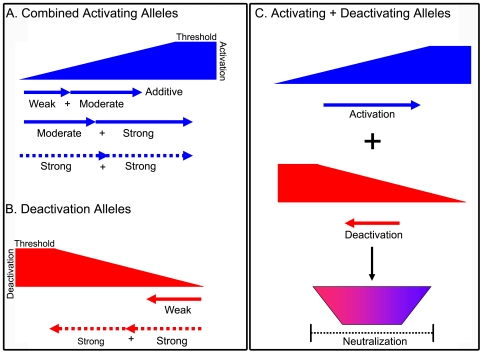
Inherited cardiomyopathy (CM) represents a diverse group of cardiac muscle diseases that present with a broad spectrum of symptoms ranging from benign to highly malignant. Contributing to this genetic complexity and clinical heterogeneity is the emergence of a cohort of patients that are double or compound heterozygotes who have inherited two different CM mutant alleles in the same or different sarcomeric gene. These patients typically have early disease onset with worse clinical outcomes. Little experimental attention has been directed towards elucidating the physiologic basis of double CM mutations at the cellular-molecular level. Here, dual gene transfer to isolated adult rat cardiac myocytes was used to determine the primary effects of co-expressing two different CM-linked mutant proteins on intact cardiac myocyte contractile physiology. Dual expression of two CM mutants, that alone moderately increase myofilament activation, tropomyosin mutant A63V and cardiac troponin mutant R146G, were shown to additively slow myocyte relaxation beyond either mutant studied in isolation. These results were qualitatively similar to a combination of moderate and strong activating CM mutant alleles alphaTmA63V and cTnI R193H, which approached a functional threshold. Interestingly, a combination of a CM myofilament deactivating mutant, troponin C G159D, together with an activating mutant, cTnIR193H, produced a hybrid phenotype that blunted the strong activating phenotype of cTnIR193H alone. This is evidence of neutralizing effects of activating/deactivating mutant alleles in combination. Taken together, this combinatorial mutant allele functional analysis lends molecular insight into disease severity and forms the foundation for a predictive model to deconstruct the myriad of possible CM double mutations in presenting patients.
Conflict of interest statement
Figures
Similar articles
-
Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants.J Mol Cell Cardiol. 2008 May;44(5):891-904. doi: 10.1016/j.yjmcc.2008.02.274. Epub 2008 Feb 26. J Mol Cell Cardiol. 2008. PMID: 18423659 Free PMC article.
-
Functional analysis of a unique troponin c mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle.Circ Heart Fail. 2009 Sep;2(5):456-64. doi: 10.1161/CIRCHEARTFAILURE.108.818237. Epub 2009 Jul 6. Circ Heart Fail. 2009. PMID: 19808376
-
Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells.Differentiation. 2012 Mar;83(3):106-15. doi: 10.1016/j.diff.2011.10.002. Epub 2011 Nov 15. Differentiation. 2012. PMID: 22364878
-
Structural interactions responsible for the assembly of the troponin complex on the muscle thin filament.Cell Struct Funct. 1997 Feb;22(1):219-23. doi: 10.1247/csf.22.219. Cell Struct Funct. 1997. PMID: 9113410 Review.
-
Calcium Ions in Inherited Cardiomyopathies.Med Chem. 2016;12(2):139-50. doi: 10.2174/157340641202160209093713. Med Chem. 2016. PMID: 26411603 Review.
Cited by
-
Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth.Circ Res. 2011 Jan 21;108(2):176-83. doi: 10.1161/CIRCRESAHA.110.231514. Epub 2010 Dec 2. Circ Res. 2011. PMID: 21127295 Free PMC article.
-
Modulating the tension-time integral of the cardiac twitch prevents dilated cardiomyopathy in murine hearts.JCI Insight. 2020 Oct 15;5(20):e142446. doi: 10.1172/jci.insight.142446. JCI Insight. 2020. PMID: 32931484 Free PMC article.
-
Cardiac Sarcomere Signaling in Health and Disease.Int J Mol Sci. 2022 Dec 19;23(24):16223. doi: 10.3390/ijms232416223. Int J Mol Sci. 2022. PMID: 36555864 Free PMC article. Review.
-
Experimental models of inherited cardiomyopathy and its therapeutics.World J Cardiol. 2014 Dec 26;6(12):1245-51. doi: 10.4330/wjc.v6.i12.1245. World J Cardiol. 2014. PMID: 25548614 Free PMC article. Review.
-
Sarcomere integrated biosensor detects myofilament-activating ligands in real time during twitch contractions in live cardiac muscle.J Mol Cell Cardiol. 2020 Oct;147:49-61. doi: 10.1016/j.yjmcc.2020.07.012. Epub 2020 Aug 11. J Mol Cell Cardiol. 2020. PMID: 32791214 Free PMC article.
References
-
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. - PubMed
-
- Braunwald E, Seidman CE, Sigwart U. Contemporary evaluation and management of hypertrophic cardiomyopathy. Circulation. 2002;106:1312–1316. - PubMed
-
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. - PubMed
-
- Doolan A, Nguyen L, Semsarian C. Hypertrophic cardiomyopathy: from “heart tumour” to a complex molecular genetic disorder. Heart Lung Circ. 2004;13:15–25. - PubMed
-
- NHLBI Program for Genomic Applications HMS. Genomics of Cardiovascular Development, Adaptation, and Remodeling. 7 A.D. June. http://www.cardiogenomics.org.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
