

Template proteogenomics: sequencing whole proteins using an imperfect database
- PMID: 20164058
- PMCID: PMC2877985
- DOI: 10.1074/mcp.M900504-MCP200
Template proteogenomics: sequencing whole proteins using an imperfect database
Abstract
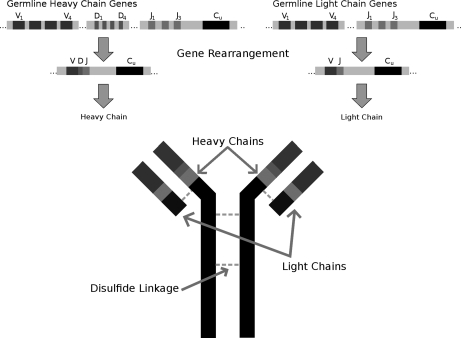
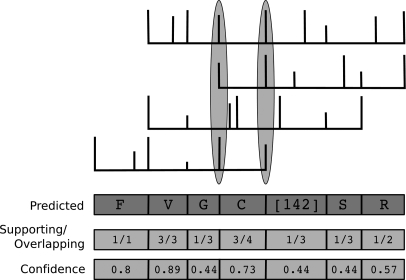
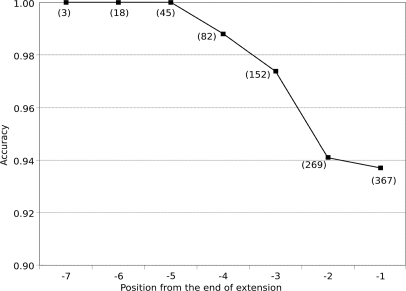
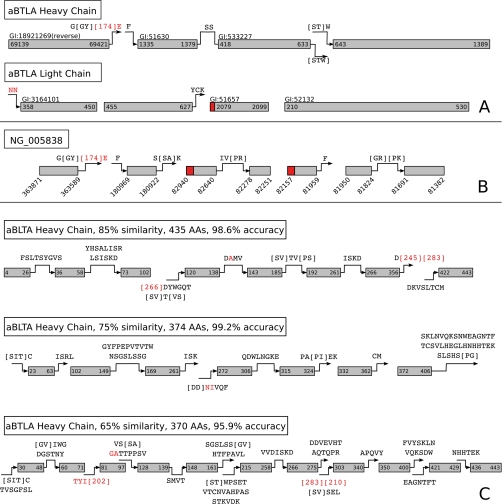
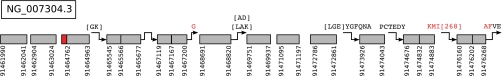
Database search algorithms are the primary workhorses for the identification of tandem mass spectra. However, these methods are limited to the identification of spectra for which peptides are present in the database, preventing the identification of peptides from mutated or alternatively spliced sequences. A variety of methods has been developed to search a spectrum against a sequence allowing for variations. Some tools determine the sequence of the homologous protein in the related species but do not report the peptide in the target organism. Other tools consider variations, including modifications and mutations, in reconstructing the target sequence. However, these tools will not work if the template (homologous peptide) is missing in the database, and they do not attempt to reconstruct the entire protein target sequence. De novo identification of peptide sequences is another possibility, because it does not require a protein database. However, the lack of database reduces the accuracy. We present a novel proteogenomic approach, GenoMS, that draws on the strengths of database and de novo peptide identification methods. Protein sequence templates (i.e. proteins or genomic sequences that are similar to the target protein) are identified using the database search tool InsPecT. The templates are then used to recruit, align, and de novo sequence regions of the target protein that have diverged from the database or are missing. We used GenoMS to reconstruct the full sequence of an antibody by using spectra acquired from multiple digests using different proteases. Antibodies are a prime example of proteins that confound standard database identification techniques. The mature antibody genes result from large-scale genome rearrangements with flexible fusion boundaries and somatic hypermutation. Using GenoMS we automatically reconstruct the complete sequences of two immunoglobulin chains with accuracy greater than 98% using a diverged protein database. Using the genome as the template, we achieve accuracy exceeding 97%.
Figures
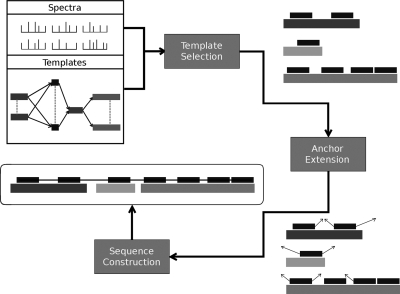
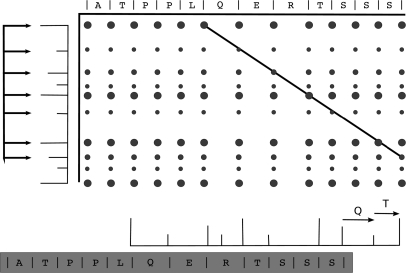
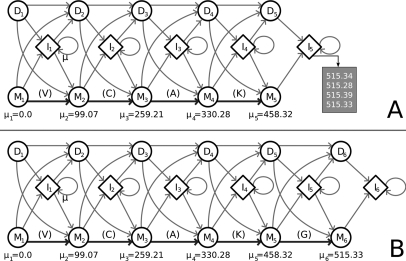
References
-
- Eng J., McCormack A., J. Y. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein data base. J. Am. Soc. Mass Spectrom 5, 976–989 - PubMed
-
- Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence data bases using mass spectrometry data. Electrophoresis 20, 3551–3567 - PubMed
-
- Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem 77, 4626–4639 - PubMed
-
- Shevchenko A., Sunyaev S., Loboda A., Shevchenko A., Bork P., Ens W., Standing K. G. (2001) Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal. Chem 73, 1917–1926 - PubMed
-
- Tsur D., Tanner S., Zandi E., Bafna V., Pevzner P. A. (2005) Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol 23, 1562–1567 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
