

How significant is a protein structure similarity with TM-score = 0.5?
- PMID: 20164152
- PMCID: PMC2913670
- DOI: 10.1093/bioinformatics/btq066
How significant is a protein structure similarity with TM-score = 0.5?
Abstract
Motivation: Protein structure similarity is often measured by root mean squared deviation, global distance test score and template modeling score (TM-score). However, the scores themselves cannot provide information on how significant the structural similarity is. Also, it lacks a quantitative relation between the scores and conventional fold classifications. This article aims to answer two questions: (i) what is the statistical significance of TM-score? (ii) What is the probability of two proteins having the same fold given a specific TM-score?
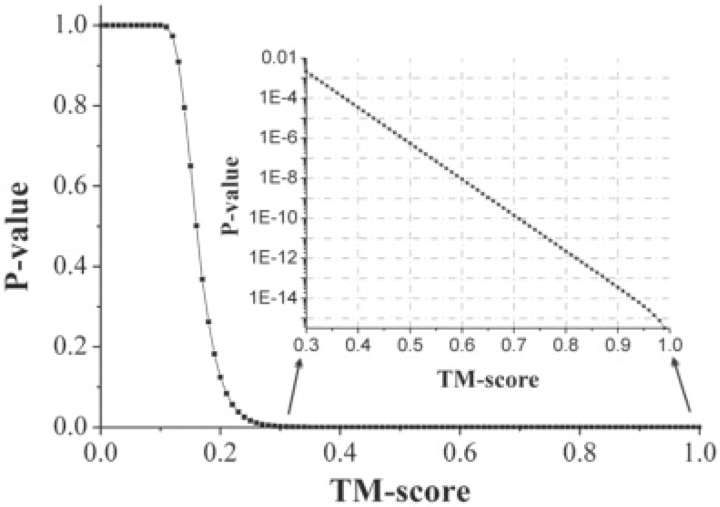
Results: We first made an all-to-all gapless structural match on 6684 non-homologous single-domain proteins in the PDB and found that the TM-scores follow an extreme value distribution. The data allow us to assign each TM-score a P-value that measures the chance of two randomly selected proteins obtaining an equal or higher TM-score. With a TM-score at 0.5, for instance, its P-value is 5.5 x 10(-7), which means we need to consider at least 1.8 million random protein pairs to acquire a TM-score of no less than 0.5. Second, we examine the posterior probability of the same fold proteins from three datasets SCOP, CATH and the consensus of SCOP and CATH. It is found that the posterior probability from different datasets has a similar rapid phase transition around TM-score=0.5. This finding indicates that TM-score can be used as an approximate but quantitative criterion for protein topology classification, i.e. protein pairs with a TM-score >0.5 are mostly in the same fold while those with a TM-score <0.5 are mainly not in the same fold.
Figures





References
-
- Ben-David M, et al. Assess ment of CASP8 structure predictions for template free targets. Proteins. 2009;77(Suppl. 9):50–65. - PubMed
-
- Berman HM, et al. The protein data bank. Acta Crystallogr., Sect D: Biol. Crystallogr. 2002;58:899–907. - PubMed
-
- Betancourt MR, Skolnick J. Universal similarity measure for comparing protein structures. Biopolymers. 2001;59:305–309. - PubMed
-
- Chothia C, et al. Evolution of the protein repertoire. Science. 2003;300:1701–1703. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
