

Reciprocal regulation of AKT and MAP kinase dictates virus-host cell fusion
- PMID: 20164223
- PMCID: PMC2863742
- DOI: 10.1128/JVI.01940-09
Reciprocal regulation of AKT and MAP kinase dictates virus-host cell fusion
Abstract
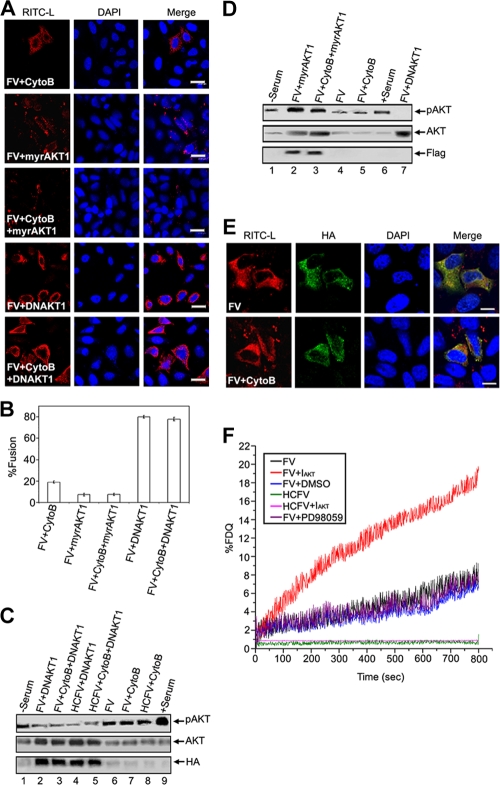
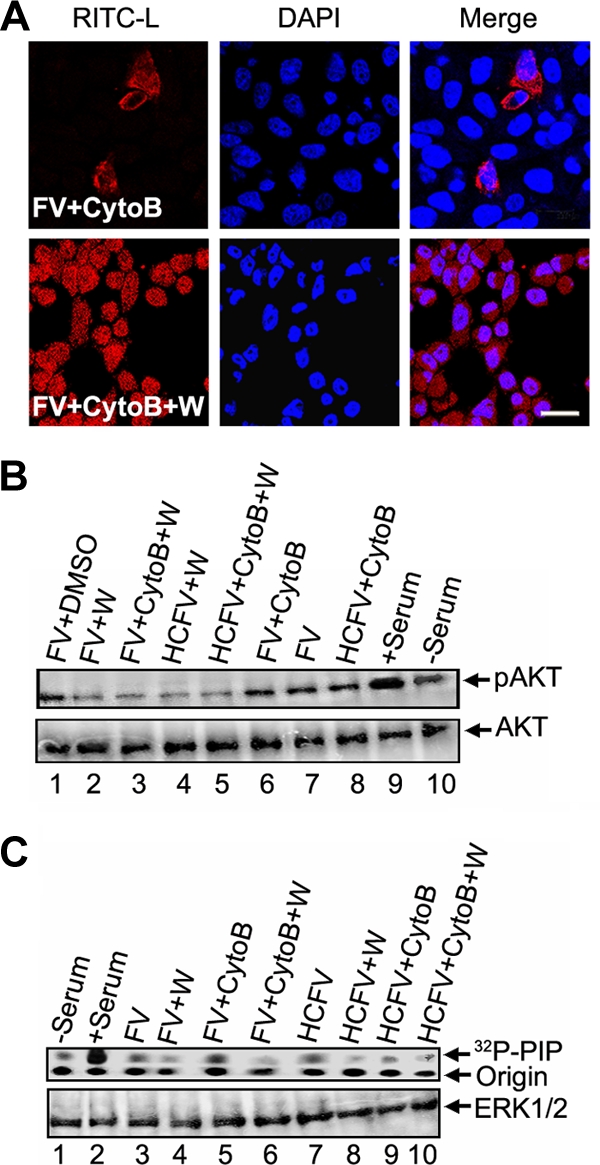
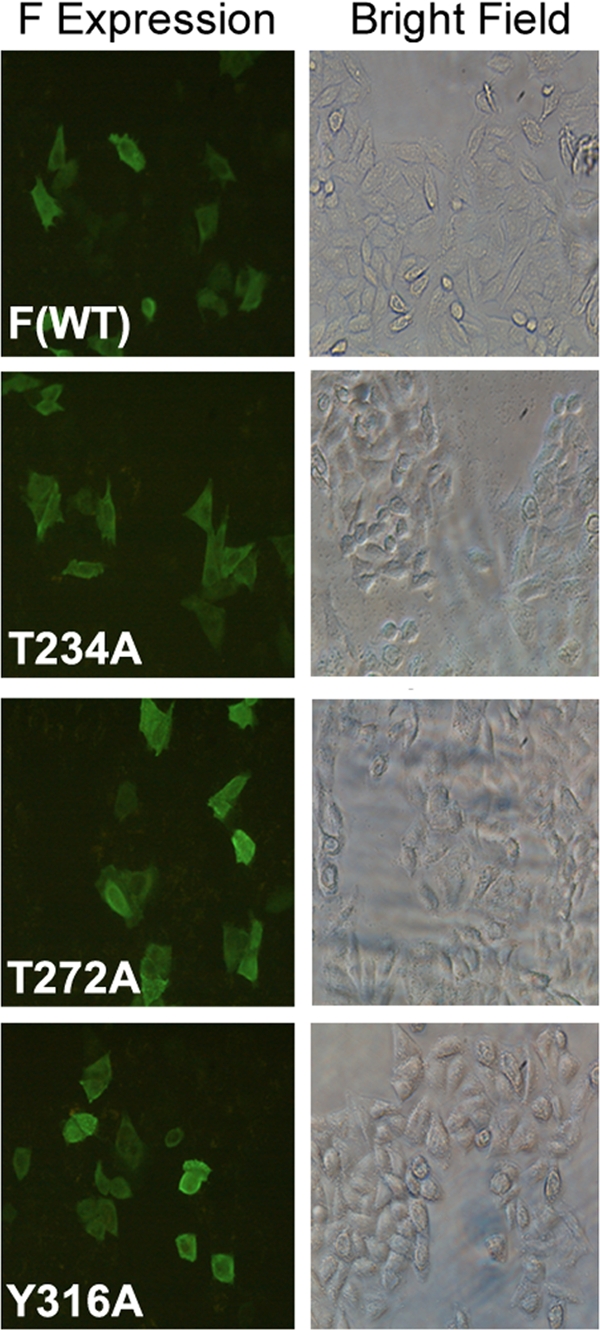
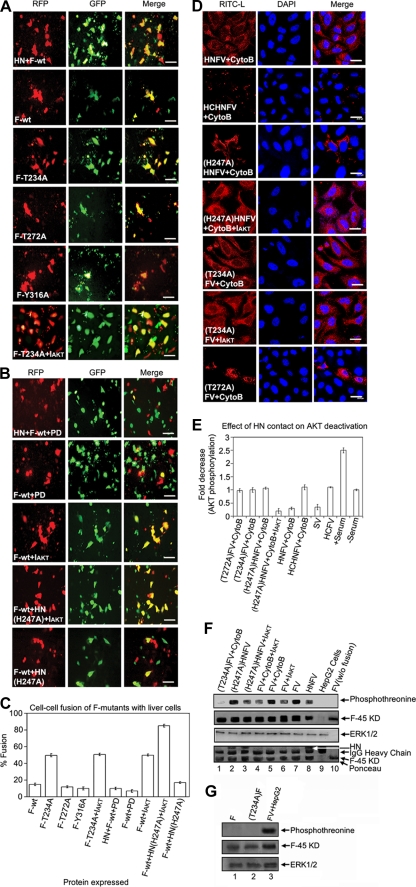
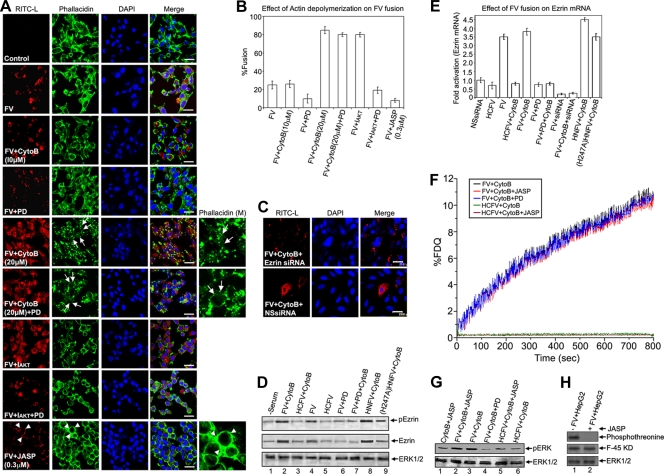
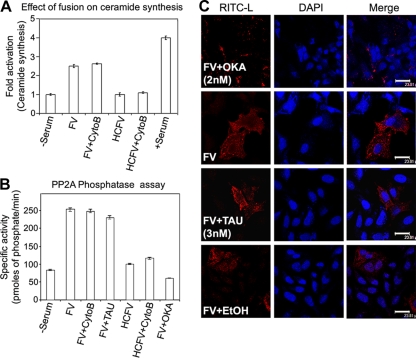
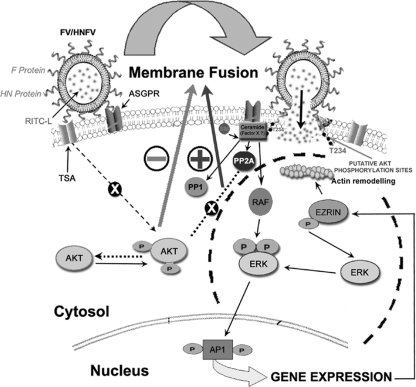
Viruses of the Paramyxoviridae family bind to their host cells by using hemagglutinin-neuraminidase (HN), which enhances fusion protein (F)-mediated membrane fusion. Although respiratory syncytial virus and parainfluenza virus 5 of this family are suggested to trigger host cell signaling during infection, the virus-induced intracellular signals dictating virus-cell fusion await elucidation. Using an F- or HN-F-containing reconstituted envelope of Sendai virus, another paramyxovirus, we revealed the role and regulation of AKT1 and Raf/MEK/ERK cascades during viral fusion with liver cells. Our observation that extracellular signal-regulated kinase (ERK) activation promotes viral fusion via ezrin-mediated cytoskeletal rearrangements, whereas AKT1 attenuates fusion by promoting phosphorylation of F protein, indicates a counteractive regulation of viral fusion by reciprocal activation of AKT1 and mitogen-activated protein kinase (MAPK) cascades, establishing a novel conceptual framework for a therapeutic strategy.
Figures

Similar articles
-
A histidine switch in hemagglutinin-neuraminidase triggers paramyxovirus-cell membrane fusion.J Virol. 2009 Feb;83(4):1727-41. doi: 10.1128/JVI.02026-08. Epub 2008 Dec 3. J Virol. 2009. PMID: 19052089 Free PMC article.
-
Recombinant Sendai viruses expressing fusion proteins with two furin cleavage sites mimic the syncytial and receptor-independent infection properties of respiratory syncytial virus.J Virol. 2011 Mar;85(6):2771-80. doi: 10.1128/JVI.02065-10. Epub 2011 Jan 12. J Virol. 2011. PMID: 21228237 Free PMC article.
-
Human parainfluenza virus 3 field strains undergo extracellular fusion protein cleavage to activate entry.mBio. 2024 Nov 13;15(11):e0232724. doi: 10.1128/mbio.02327-24. Epub 2024 Oct 9. mBio. 2024. PMID: 39382296 Free PMC article.
-
Paramyxovirus membrane fusion: lessons from the F and HN atomic structures.Virology. 2006 Jan 5;344(1):30-7. doi: 10.1016/j.virol.2005.09.007. Virology. 2006. PMID: 16364733 Free PMC article. Review.
-
Membrane uncoating of intact enveloped viruses.J Virol. 2010 Nov;84(21):10946-55. doi: 10.1128/JVI.00229-10. Epub 2010 Jul 28. J Virol. 2010. PMID: 20668081 Free PMC article. Review.
Cited by
-
Cholesterol dependence of Newcastle Disease Virus entry.Biochim Biophys Acta. 2012 Mar;1818(3):753-61. doi: 10.1016/j.bbamem.2011.12.004. Epub 2011 Dec 13. Biochim Biophys Acta. 2012. PMID: 22192779 Free PMC article.
-
Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2.J Virol. 2012 Feb;86(3):1339-47. doi: 10.1128/JVI.06096-11. Epub 2011 Nov 16. J Virol. 2012. PMID: 22090124 Free PMC article.
-
Sendai virus recruits cellular villin to remodel actin cytoskeleton during fusion with hepatocytes.Mol Biol Cell. 2017 Dec 15;28(26):3801-3814. doi: 10.1091/mbc.E17-06-0400. Epub 2017 Oct 26. Mol Biol Cell. 2017. PMID: 29074568 Free PMC article.
-
The MAPK Response to Virus Infection Is Modified by Probenecid.Curr Issues Mol Biol. 2025 Apr 2;47(4):246. doi: 10.3390/cimb47040246. Curr Issues Mol Biol. 2025. PMID: 40699645 Free PMC article. Review.
-
1-Formyl-β-carboline Derivatives Block Newcastle Disease Virus Proliferation through Suppressing Viral Adsorption and Entry Processes.Biomolecules. 2021 Nov 12;11(11):1687. doi: 10.3390/biom11111687. Biomolecules. 2021. PMID: 34827684 Free PMC article.
References
-
- Avota, E., A. Avots, S. Niewiesk, L. P. Kane, U. Bommhardt, V. ter Meulen, and S. Schneider-Schaulies. 2001. Disruption of Akt kinase activation is important for immunosuppression induced by measles virus. Nat. Med. 7:725-731. - PubMed
-
- Bagai, S., and D. P. Sarkar. 1994. Fusion-mediated microinjection of lysozyme into HepG2 cells through hemagglutinin neuraminidase-depleted Sendai virus envelopes. J. Biol. Chem. 269:1966-1972. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
