

Identification of dynamical hinge points of the L1 ligase molecular switch
- PMID: 20167653
- PMCID: PMC2844624
- DOI: 10.1261/rna.1897810
Identification of dynamical hinge points of the L1 ligase molecular switch
Abstract
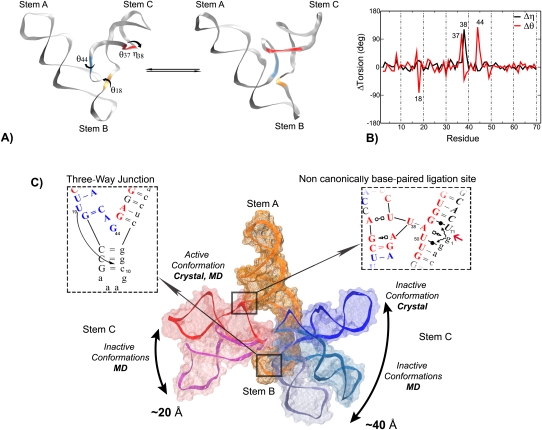
The L1 ligase is an in vitro selected ribozyme that uses a noncanonically base-paired ligation site to catalyze regioselectively and regiospecifically the 5' to 3' phosphodiester bond ligation, a reaction relevant to origin of life hypotheses that invoke an RNA world scenario. The L1 ligase crystal structure revealed two different conformational states that were proposed to represent the active and inactive forms. It remains an open question as to what degree these two conformers persist as stable conformational intermediates in solution, and along what pathway are they able to interconvert. To explore these questions, we have performed a series of molecular dynamics simulations in explicit solvent of the inactive-active conformational switch in L1 ligase. Four simulations were performed departing from both conformers in both the reactant and product states, in addition to a simulation where local unfolding in the active state was induced. From these simulations, along with crystallographic data, a set of four virtual torsion angles that span two evolutionarily conserved and restricted regions were identified as dynamical hinge points in the conformational switch transition. The ligation site visits three distinct states characterized by hydrogen bond patterns that are correlated with the formation of specific contacts that may promote catalysis. The insights gained from these simulations contribute to a more detailed understanding of the coupled catalytic/conformational switch mechanism of L1 ligase that may facilitate the design and engineering of new catalytic riboswitches.
Figures

Similar articles
-
Mapping L1 ligase ribozyme conformational switch.J Mol Biol. 2012 Oct 12;423(1):106-22. doi: 10.1016/j.jmb.2012.06.035. Epub 2012 Jul 3. J Mol Biol. 2012. PMID: 22771572 Free PMC article.
-
The structural basis of ribozyme-catalyzed RNA assembly.Science. 2007 Mar 16;315(5818):1549-53. doi: 10.1126/science.1136231. Science. 2007. PMID: 17363667
-
Evolutionary optimization of a modular ligase ribozyme: a small catalytic unit and a hairpin motif masking an element that could form an inactive structure.Nucleic Acids Res. 2010 Jun;38(10):3328-39. doi: 10.1093/nar/gkq018. Epub 2010 Jan 27. Nucleic Acids Res. 2010. PMID: 20110262 Free PMC article.
-
Crystallographic structures of the hammerhead ribozyme: relationship to ribozyme folding and catalysis.Annu Rev Biophys Biomol Struct. 1998;27:475-502. doi: 10.1146/annurev.biophys.27.1.475. Annu Rev Biophys Biomol Struct. 1998. PMID: 9646875 Review.
-
Catalytic strategies of self-cleaving ribozymes.Acc Chem Res. 2008 Aug;41(8):1027-35. doi: 10.1021/ar800050c. Epub 2008 Jul 25. Acc Chem Res. 2008. PMID: 18652494 Review.
Cited by
-
Multiscale methods for computational RNA enzymology.Methods Enzymol. 2015;553:335-74. doi: 10.1016/bs.mie.2014.10.064. Epub 2015 Jan 22. Methods Enzymol. 2015. PMID: 25726472 Free PMC article.
-
A new way to see RNA.Q Rev Biophys. 2011 Nov;44(4):433-66. doi: 10.1017/S0033583511000059. Epub 2011 May 18. Q Rev Biophys. 2011. PMID: 21729350 Free PMC article.
-
Mapping L1 ligase ribozyme conformational switch.J Mol Biol. 2012 Oct 12;423(1):106-22. doi: 10.1016/j.jmb.2012.06.035. Epub 2012 Jul 3. J Mol Biol. 2012. PMID: 22771572 Free PMC article.
-
AMIGOS III: pseudo-torsion angle visualization and motif-based structure comparison of nucleic acids.Bioinformatics. 2022 May 13;38(10):2937-2939. doi: 10.1093/bioinformatics/btac207. Bioinformatics. 2022. PMID: 35561202 Free PMC article.
-
The impact of a ligand binding on strand migration in the SAM-I riboswitch.PLoS Comput Biol. 2013;9(5):e1003069. doi: 10.1371/journal.pcbi.1003069. Epub 2013 May 16. PLoS Comput Biol. 2013. PMID: 23704854 Free PMC article.
References
-
- Allen M, Tildesley D. Computer simulation of liquids. Oxford University Press; Oxford, UK: 1987.
-
- Bartel DP, Szostak JW. Isolation of new ribozymes from a large pool of random sequences. Science. 1993;261:1411–1418. - PubMed
-
- Bokinsky G, Zhuang X. Single-molecule RNA folding. Acc Chem Res. 2005;38:566–573. - PubMed
-
- Bunka DHJ, Stockley PG. Aptamers come of age—at last. Nat Rev Microbiol. 2006;4:588–596. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources