

AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes
- PMID: 20167927
- PMCID: PMC2920052
- DOI: 10.1161/CIRCRESAHA.109.212530
AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes
Abstract
Rationale: AMP-activated protein kinase (AMPK) is an energy sensor and ubiquitously expressed in vascular cells. Recent studies suggest that AMPK activation improves endothelial function by counteracting oxidative stress in endothelial cells. How AMPK suppresses oxidative stress remains to be established.
Objective: The aim of this study is to examine the effects of AMPK in regulating NAD(P)H oxidase, oxidative stress, and endothelial function.
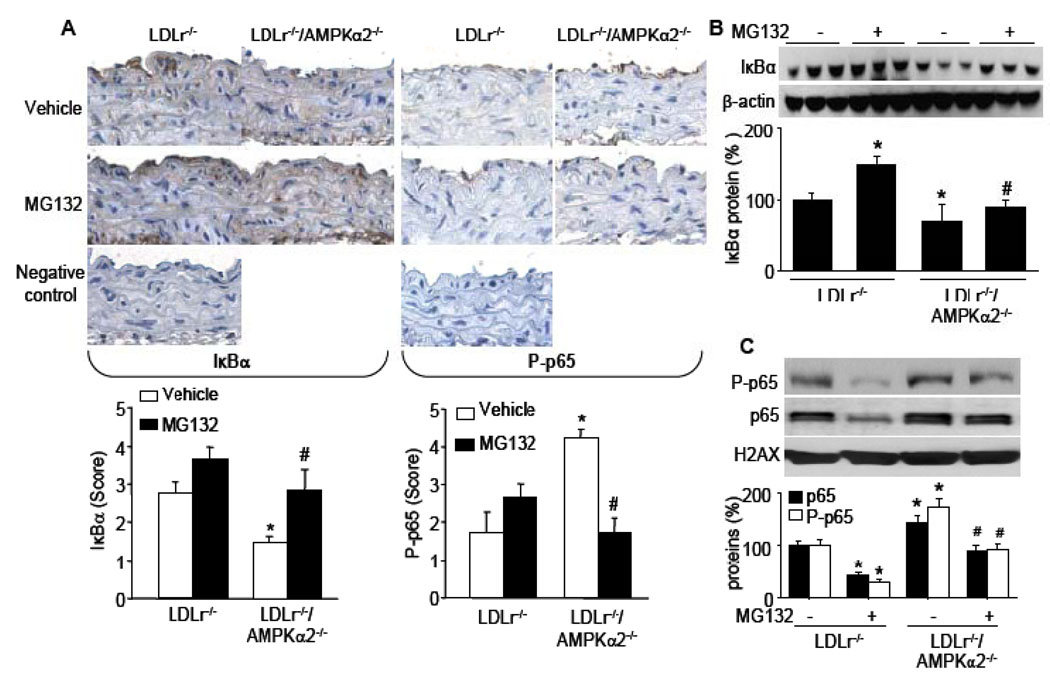
Methods and results: The markers of oxidative stress, NAD(P)H oxidase subunit expression (gp91(phox), p47(phox), p67(phox), NOX1 to -4), NAD(P)H oxidase-mediated superoxide production, 26S proteasome activity, IkappaBalpha degradation, and nuclear translocation of nuclear factor (NF)-kappaB (p50 and p65) were examined in cultured human umbilical vein endothelial cells and mouse aortas isolated from AMPKalpha2 deficient mice. Compared to the wild type, acetylcholine-induced endothelium-dependent relaxation was significantly impaired in parallel with increased production of oxidants in AMPKalpha2(-/-) mice. Further, pretreatment of aorta with either superoxide dismutase (SOD) or tempol or apocynin significantly improved acetylcholine-induced endothelium-dependent relaxation in AMPKalpha2(-/-) mice. Analysis of aortic endothelial cells from AMPKalpha2(-/-) mice and human umbilical vein endothelial cells expressing dominant negative AMPK or AMPKalpha2-specific siRNA revealed that loss of AMPK activity increased NAD(P)H oxidase subunit expression (gp91(phox), p47(phox), p67(phox), NOX1 and -4), NAD(P)H oxidase-mediated superoxide production, 26S proteasome activity, IkappaBalpha degradation, and nuclear translocation of NF-kappaB (p50 and p65), whereas AMPK activation by AICAR or overexpression of constitutively active AMPK had the opposite effect. Consistently, we found that genetic deletion of AMPKalpha2 in low-density lipoprotein receptor knockout (LDLr(-/-)) strain markedly increased 26S proteasome activity, IkappaB degradation, NF-kappaB transactivation, NAD(P)H oxidase subunit overexpression, oxidative stress, and endothelial dysfunction, all of which were largely suppressed by chronic administration of MG132, a potent cell permeable proteasome inhibitor.
Conclusions: We conclude that AMPKalpha2 functions as a physiological suppressor of NAD(P)H oxidase and ROS production in endothelial cells. In this way, AMPK maintains the nonatherogenic and noninflammatory phenotype of endothelial cells.
Figures







Similar articles
-
Adenosine monophosphate activated protein kinase regulates ABCG1-mediated oxysterol efflux from endothelial cells and protects against hypercholesterolemia-induced endothelial dysfunction.Arterioscler Thromb Vasc Biol. 2010 Jul;30(7):1354-62. doi: 10.1161/ATVBAHA.110.204230. Epub 2010 Apr 15. Arterioscler Thromb Vasc Biol. 2010. PMID: 20395595
-
CD4+ T lymphocytes mediate hypercholesterolemia-induced endothelial dysfunction via a NAD(P)H oxidase-dependent mechanism.Am J Physiol Heart Circ Physiol. 2008 Jun;294(6):H2619-26. doi: 10.1152/ajpheart.00989.2007. Epub 2008 Apr 11. Am J Physiol Heart Circ Physiol. 2008. PMID: 18408127
-
Activation of NF-kappaB by palmitate in endothelial cells: a key role for NADPH oxidase-derived superoxide in response to TLR4 activation.Arterioscler Thromb Vasc Biol. 2009 Sep;29(9):1370-5. doi: 10.1161/ATVBAHA.109.188813. Epub 2009 Jun 18. Arterioscler Thromb Vasc Biol. 2009. PMID: 19542021 Free PMC article.
-
NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology.Antioxid Redox Signal. 2009 Apr;11(4):791-810. doi: 10.1089/ars.2008.2220. Antioxid Redox Signal. 2009. PMID: 18783313 Free PMC article. Review.
-
Targeting NADPH oxidases in vascular pharmacology.Vascul Pharmacol. 2012 May-Jun;56(5-6):216-31. doi: 10.1016/j.vph.2012.02.012. Epub 2012 Mar 3. Vascul Pharmacol. 2012. PMID: 22405985 Free PMC article. Review.
Cited by
-
Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission.Diabetes. 2017 Jan;66(1):193-205. doi: 10.2337/db16-0915. Epub 2016 Oct 13. Diabetes. 2017. PMID: 27737949 Free PMC article.
-
A novel inverse relationship between metformin-triggered AMPK-SIRT1 signaling and p53 protein abundance in high glucose-exposed HepG2 cells.Am J Physiol Cell Physiol. 2012 Jul 1;303(1):C4-C13. doi: 10.1152/ajpcell.00296.2011. Epub 2012 Feb 29. Am J Physiol Cell Physiol. 2012. PMID: 22378745 Free PMC article.
-
Analysis of Differentially Expressed Genes in Endothelial Cells Following Tumor Cell Adhesion, and the Role of PRKAA2 and miR-124-3p.Front Cell Dev Biol. 2021 Feb 19;9:604038. doi: 10.3389/fcell.2021.604038. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33681194 Free PMC article.
-
Adiponectin inhibits NLRP3 inflammasome by modulating the AMPK-ROS pathway.Int J Clin Exp Pathol. 2018 Jul 1;11(7):3338-3347. eCollection 2018. Int J Clin Exp Pathol. 2018. PMID: 31949710 Free PMC article.
-
Epithelial sodium channels in endothelial cells mediate diet-induced endothelium stiffness and impaired vascular relaxation in obese female mice.Metabolism. 2019 Oct;99:57-66. doi: 10.1016/j.metabol.2019.153946. Epub 2019 Jul 11. Metabolism. 2019. PMID: 31302199 Free PMC article.
References
-
- Schulz E, Anter E, Zou MH, Keaney JF., Jr Estradiol-mediated endothelial nitric oxide synthase association with heat shock protein 90 requires adenosine monophosphate-dependent protein kinase. Circulation. 2005;111:3473–3480. - PubMed
-
- Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. - PubMed
-
- Zou MH, Hou XY, Shi CM, Nagata D, Walsh K, Cohen RA. Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem. 2002;277:32552–32557. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
