

Eicosanoids and cancer
- PMID: 20168319
- PMCID: PMC2898136
- DOI: 10.1038/nrc2809
Eicosanoids and cancer
Abstract
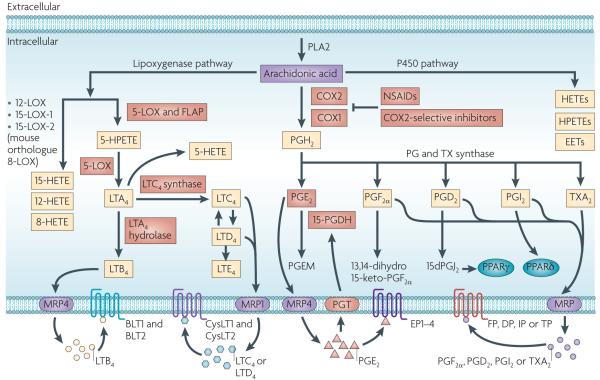
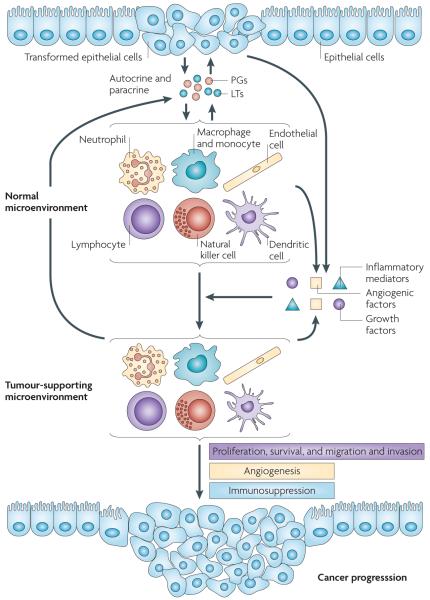
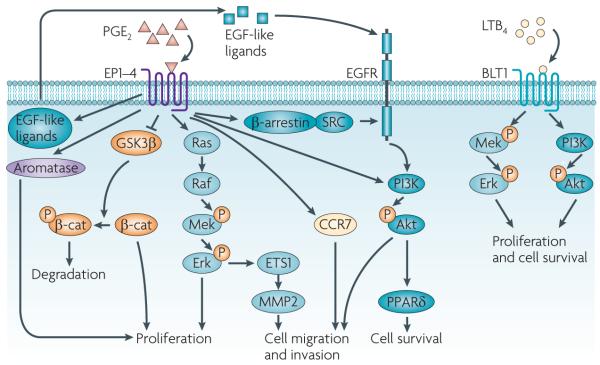
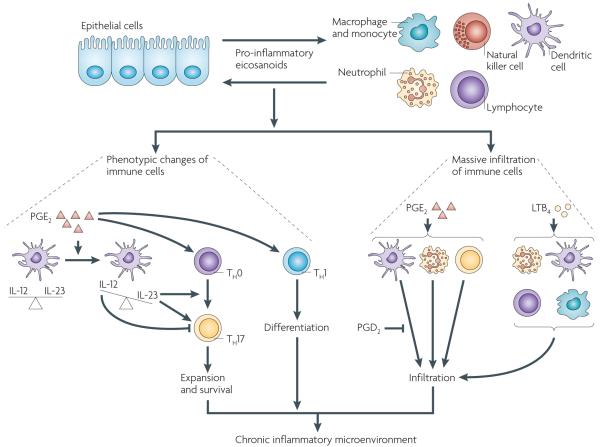
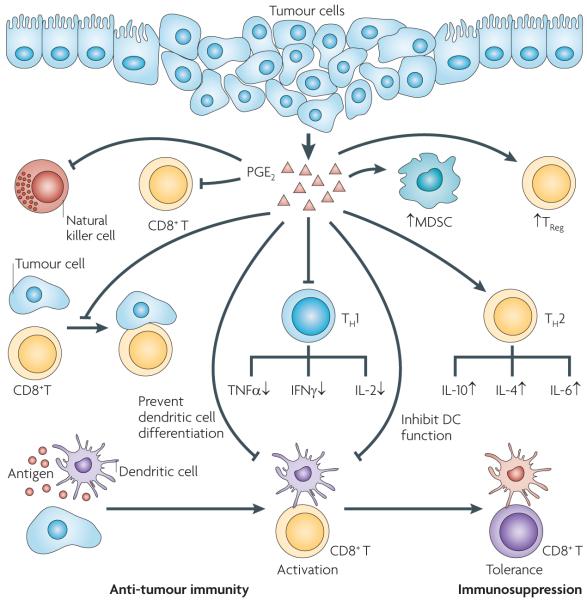
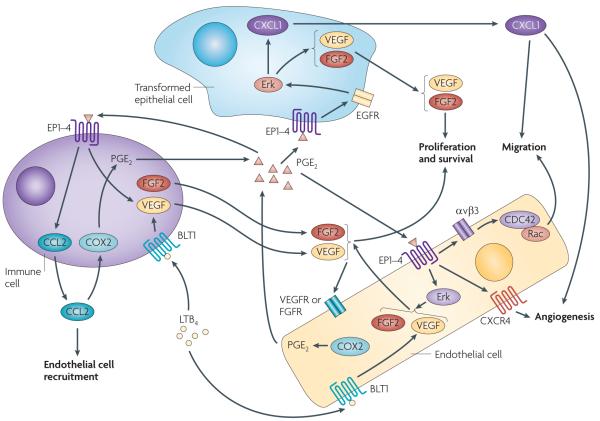
Eicosanoids, including prostaglandins and leukotrienes, are biologically active lipids that have been implicated in various pathological processes, such as inflammation and cancer. This Review highlights our understanding of the intricate roles of eicosanoids in epithelial-derived tumours and their microenvironment. The knowledge of how these lipids orchestrate the complex interactions between transformed epithelial cells and the surrounding stromal cells is crucial for understanding tumour evolution, progression and metastasis. Understanding the molecular mechanisms underlying the role of prostaglandins and other eicosanoids in cancer progression will help to develop more effective cancer chemopreventive and/or therapeutic agents.
Figures
References
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. - PubMed
-
- Ting AH, McGarvey KM, Baylin SB. The cancer epigenome — components and functional correlates. Genes Dev. 2006;20:3215–3231. - PubMed
-
- Woutersen RA, Appel MJ, van Garderen-Hoetmer A, Wijnands MV. Dietary fat and carcinogenesis. Mutat. Res. 1999;443:111–127. - PubMed
-
- Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17:55–67. - PubMed
-
- Pidgeon GP, et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Rev. 2007;26:503–524. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
