

Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion
- PMID: 20169190
- PMCID: PMC2820536
- DOI: 10.1371/journal.ppat.1000777
Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion
Abstract
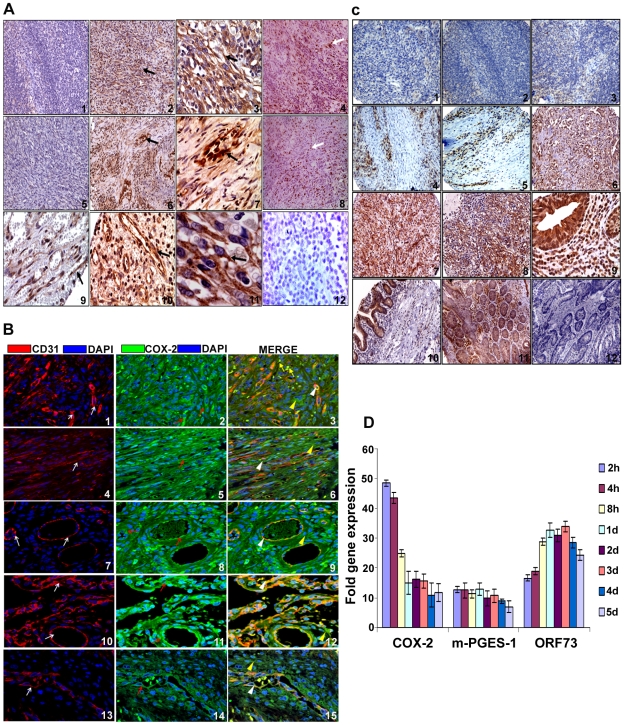
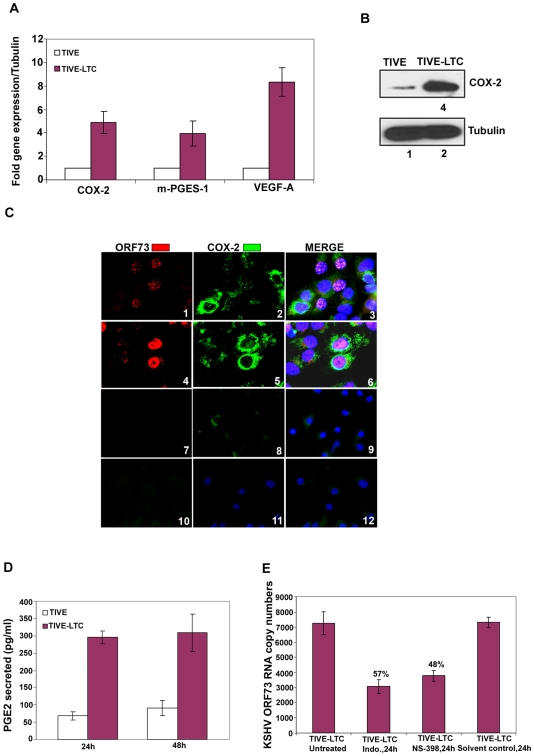
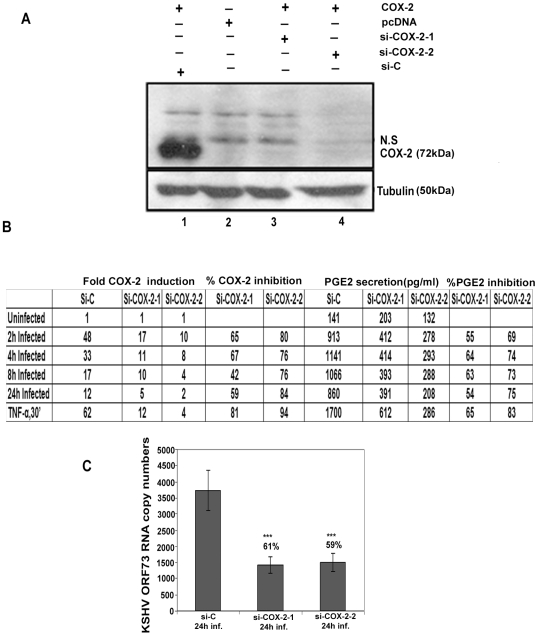
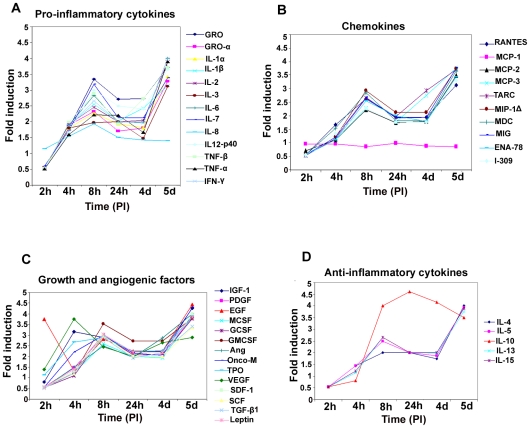
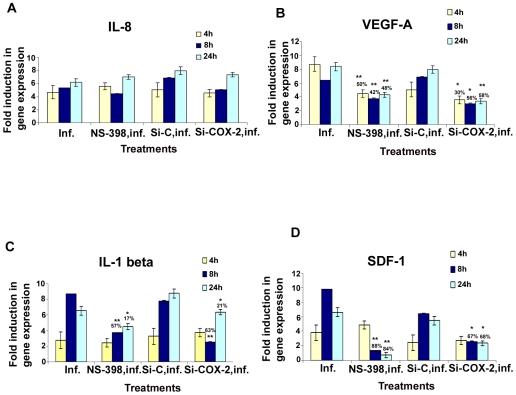
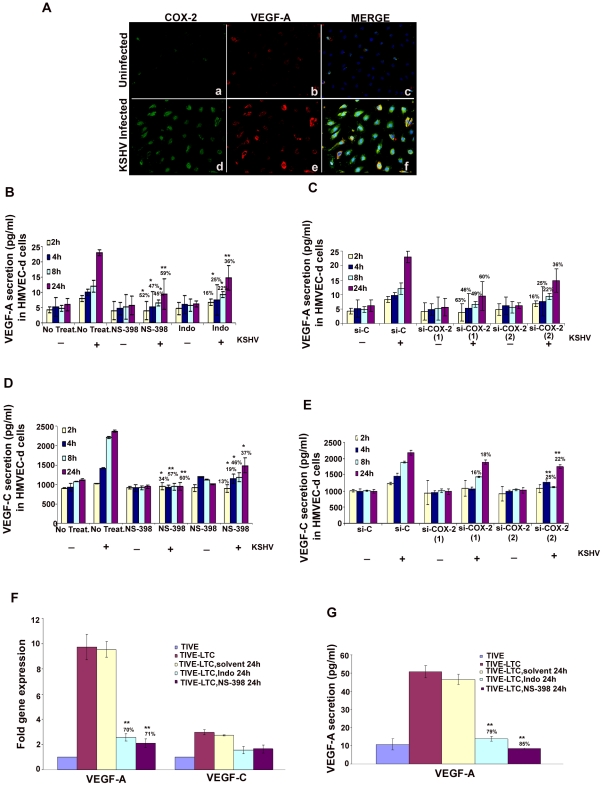
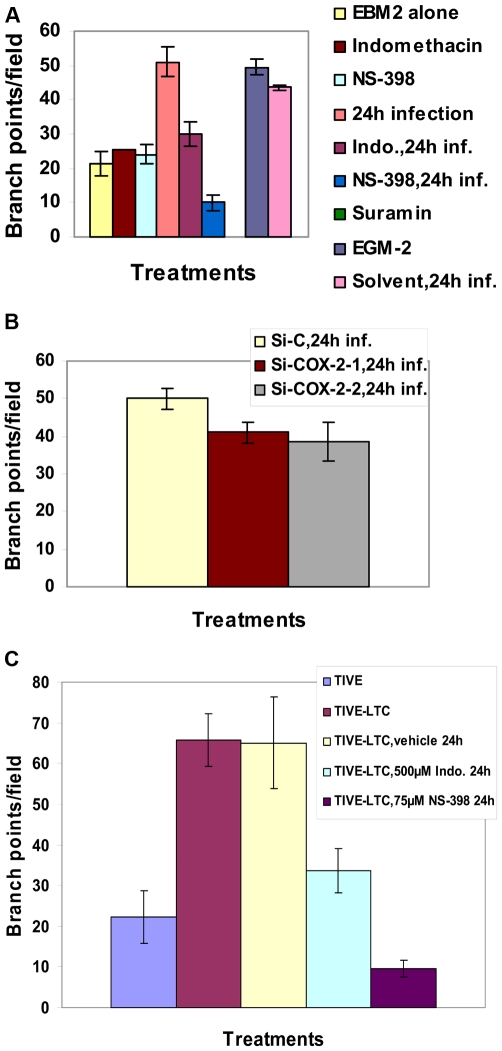
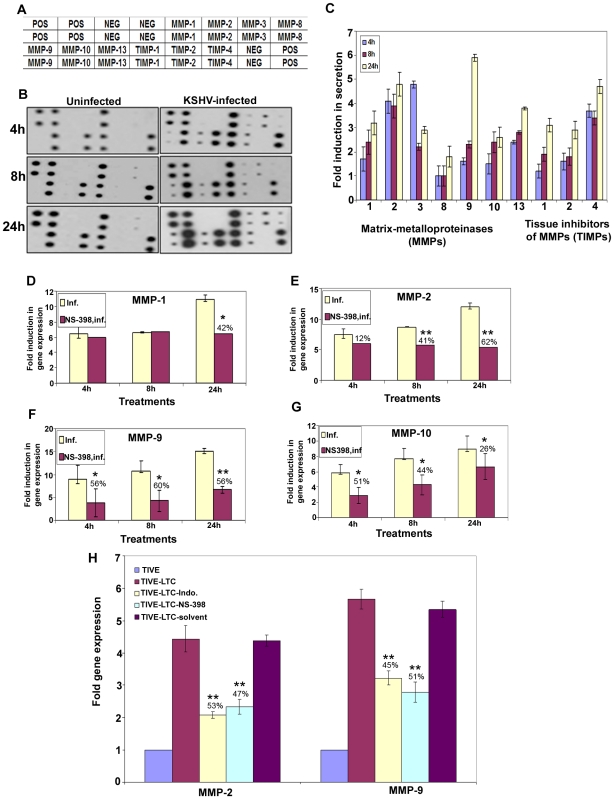
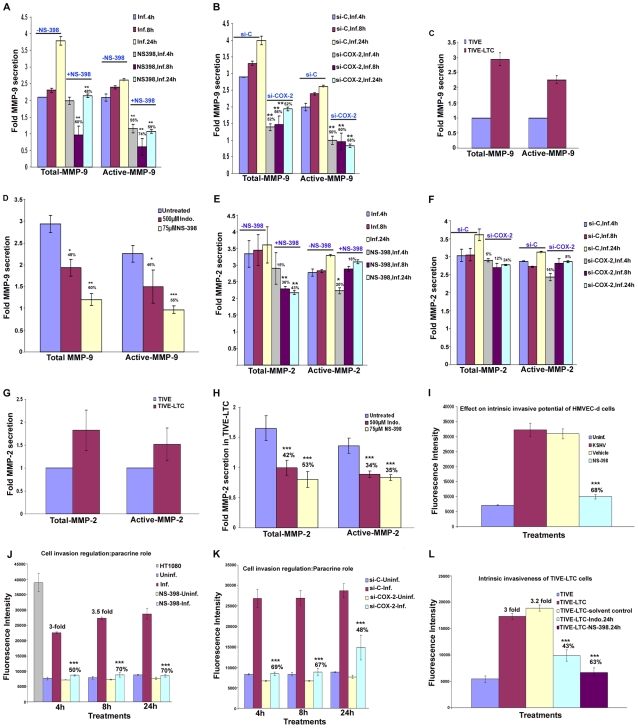
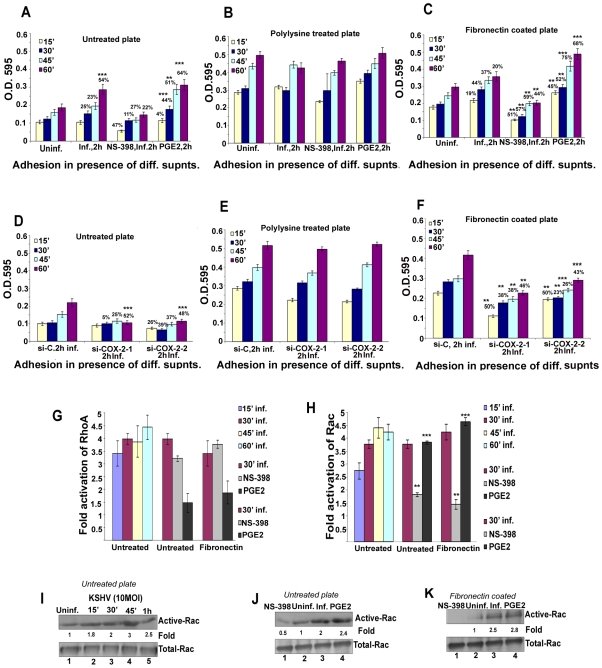
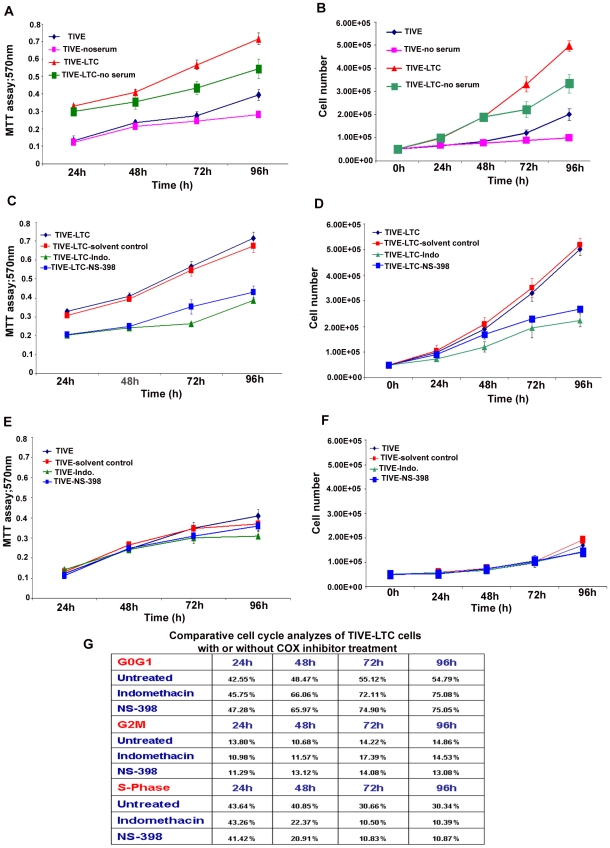
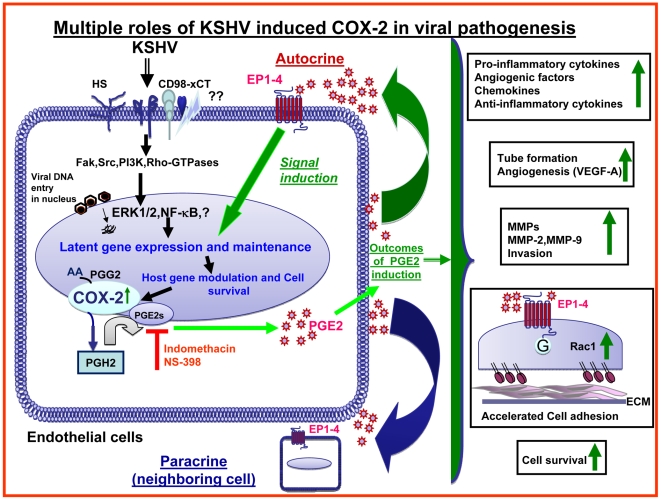
Kaposi's sarcoma (KS), an enigmatic endothelial cell vascular neoplasm, is characterized by the proliferation of spindle shaped endothelial cells, inflammatory cytokines (ICs), growth factors (GFs) and angiogenic factors. KSHV is etiologically linked to KS and expresses its latent genes in KS lesion endothelial cells. Primary infection of human micro vascular endothelial cells (HMVEC-d) results in the establishment of latent infection and reprogramming of host genes, and cyclooxygenase-2 (COX-2) is one of the highly up-regulated genes. Our previous study suggested a role for COX-2 in the establishment and maintenance of KSHV latency. Here, we examined the role of COX-2 in the induction of ICs, GFs, angiogenesis and invasive events occurring during KSHV de novo infection of endothelial cells. A significant amount of COX-2 was detected in KS tissue sections. Telomerase-immortalized human umbilical vein endothelial cells supporting KSHV stable latency (TIVE-LTC) expressed elevated levels of functional COX-2 and microsomal PGE2 synthase (m-PGES), and secreted the predominant eicosanoid inflammatory metabolite PGE2. Infected HMVEC-d and TIVE-LTC cells secreted a variety of ICs, GFs, angiogenic factors and matrix metalloproteinases (MMPs), which were significantly abrogated by COX-2 inhibition either by chemical inhibitors or by siRNA. The ability of these factors to induce tube formation of uninfected endothelial cells was also inhibited. PGE2, secreted early during KSHV infection, profoundly increased the adhesion of uninfected endothelial cells to fibronectin by activating the small G protein Rac1. COX-2 inhibition considerably reduced KSHV latent ORF73 gene expression and survival of TIVE-LTC cells. Collectively, these studies underscore the pivotal role of KSHV induced COX-2/PGE2 in creating KS lesion like microenvironment during de novo infection. Since COX-2 plays multiple roles in KSHV latent gene expression, which themselves are powerful mediators of cytokine induction, anti-apoptosis, cell survival and viral genome maintainence, effective inhibition of COX-2 via well-characterized clinically approved COX-2 inhibitors could potentially be used in treatment to control latent KSHV infection and ameliorate KS.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Kaposi's Sarcoma-Associated Herpesvirus Infection Induces the Expression of Neuroendocrine Genes in Endothelial Cells.J Virol. 2020 Mar 31;94(8):e01692-19. doi: 10.1128/JVI.01692-19. Print 2020 Mar 31. J Virol. 2020. PMID: 31969437 Free PMC article.
-
Cyclooxygenase 2 induced by Kaposi's sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression.J Virol. 2006 Jul;80(13):6534-52. doi: 10.1128/JVI.00231-06. J Virol. 2006. PMID: 16775340 Free PMC article.
-
Cyclooxygenase-2-prostaglandin E2-eicosanoid receptor inflammatory axis: a key player in Kaposi's sarcoma-associated herpes virus associated malignancies.Transl Res. 2013 Aug;162(2):77-92. doi: 10.1016/j.trsl.2013.03.004. Epub 2013 Apr 6. Transl Res. 2013. PMID: 23567332 Free PMC article. Review.
-
Kaposi's sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis.J Virol. 2009 Apr;83(7):3342-64. doi: 10.1128/JVI.02052-08. Epub 2009 Jan 21. J Virol. 2009. PMID: 19158252 Free PMC article.
-
Endothelial cell- and lymphocyte-based in vitro systems for understanding KSHV biology.Curr Top Microbiol Immunol. 2007;312:211-44. doi: 10.1007/978-3-540-34344-8_8. Curr Top Microbiol Immunol. 2007. PMID: 17089799 Review.
Cited by
-
EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination.PLoS Pathog. 2013;9(7):e1003510. doi: 10.1371/journal.ppat.1003510. Epub 2013 Jul 18. PLoS Pathog. 2013. PMID: 23874206 Free PMC article.
-
Kaposi's sarcoma-associated herpesvirus-induced angiogenin plays roles in latency via the phospholipase C gamma pathway: blocking angiogenin inhibits latent gene expression and induces the lytic cycle.J Virol. 2011 Mar;85(6):2666-85. doi: 10.1128/JVI.01532-10. Epub 2011 Jan 5. J Virol. 2011. PMID: 21209106 Free PMC article.
-
Inflammatory stress and sarcomagenesis: a vicious interplay.Cell Stress Chaperones. 2014 Jan;19(1):1-13. doi: 10.1007/s12192-013-0449-4. Epub 2013 Aug 27. Cell Stress Chaperones. 2014. PMID: 24046208 Free PMC article. Review.
-
Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases.Microorganisms. 2020 Mar 11;8(3):388. doi: 10.3390/microorganisms8030388. Microorganisms. 2020. PMID: 32168836 Free PMC article. Review.
-
Arachidonic Acid Derived Lipid Mediators Influence Kaposi's Sarcoma-Associated Herpesvirus Infection and Pathogenesis.Front Microbiol. 2019 Mar 12;10:358. doi: 10.3389/fmicb.2019.00358. eCollection 2019. Front Microbiol. 2019. PMID: 30915039 Free PMC article. Review.
References
-
- Boshoff C, Weiss RA. Kaposi's sarcoma-associated herpesvirus. Adv Cancer Res. 1998;75:57–86. - PubMed
-
- Fidler IJ, Singh RK, Yoneda J, Kumar R, Xu L, et al. Critical determinants of neoplastic angiogenesis. Cancer J. 2000;6(Suppl 3):S225–236. - PubMed
-
- Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. - PubMed
-
- Vart RJ, Nikitenko LL, Lagos D, Trotter MW, Cannon M, et al. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 2007;67:4042–4051. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
