

Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity
- PMID: 20173741
- PMCID: PMC3058354
- DOI: 10.1038/ncb2023
Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity
Abstract
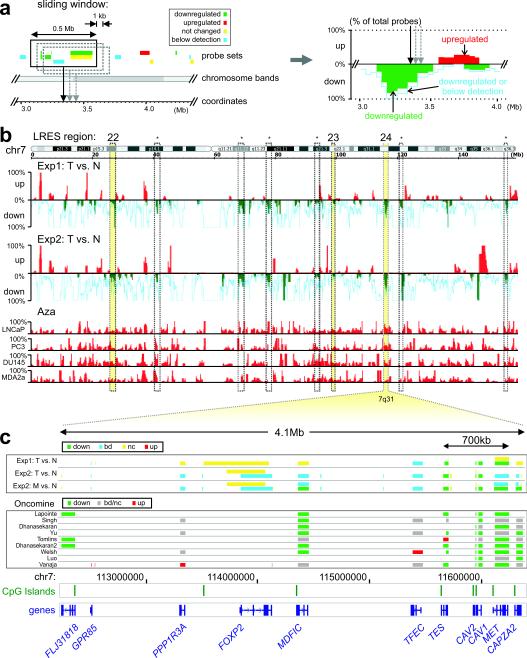
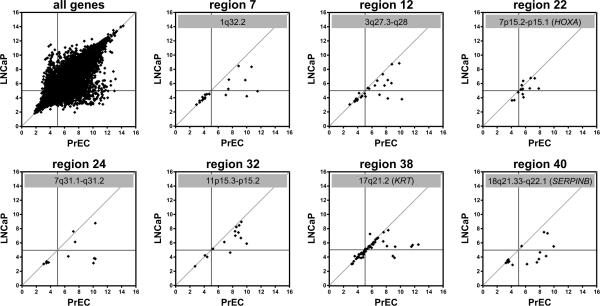
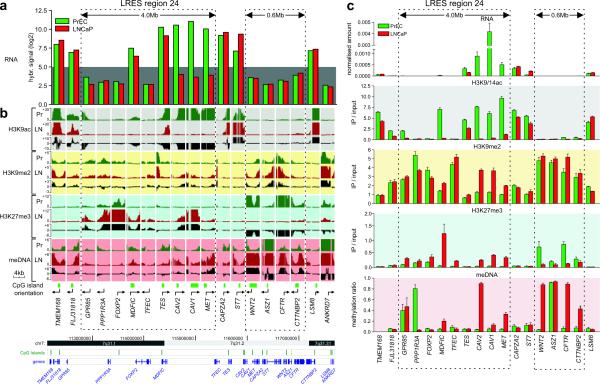
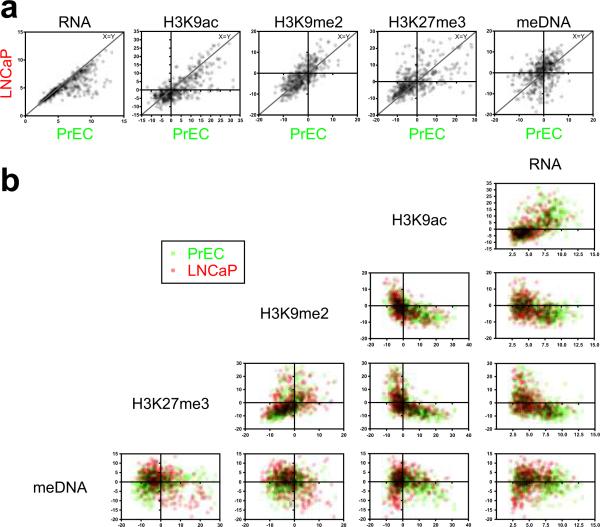
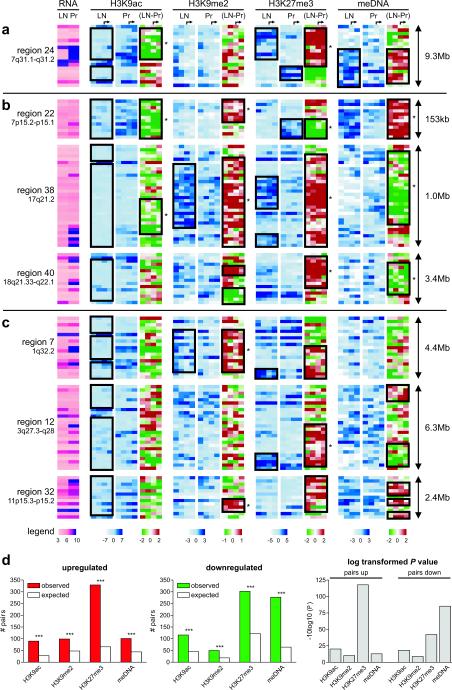
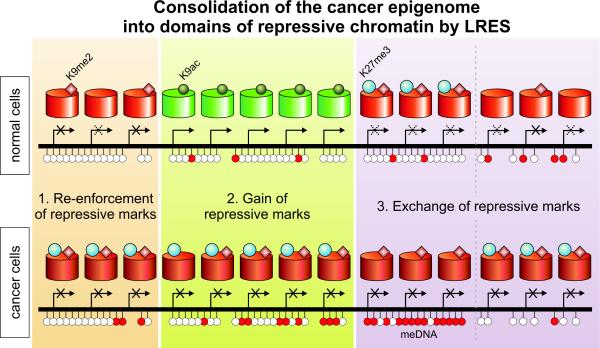
Silencing of individual genes can occur by genetic and epigenetic processes during carcinogenesis, but the underlying mechanisms remain unclear. By creating an integrated prostate cancer epigenome map using tiling arrays, we show that contiguous regions of gene suppression commonly occur through long-range epigenetic silencing (LRES). We identified 47 LRES regions in prostate cancer, typically spanning about 2 Mb and harbouring approximately 12 genes, with a prevalence of tumour suppressor and miRNA genes. Our data reveal that LRES is associated with regional histone deacetylation combined with subdomains of different epigenetic remodelling patterns, which include re-enforcement, gain or exchange of repressive histone, and DNA methylation marks. The transcriptional and epigenetic state of genes in normal prostate epithelial and human embryonic stem cells can play a critical part in defining the mode of cancer-associated epigenetic remodelling. We propose that consolidation or effective reduction of the cancer genome commonly occurs in domains through a combination of LRES and LOH or genomic deletion, resulting in reduced transcriptional plasticity within these regions.
Figures

References
-
- Baylin SB, et al. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Human molecular genetics. 2001;10:687–692. - PubMed
-
- Bird AP, Wolffe AP. Methylation-induced repression - Belts, braces, and chromatin. Cell. 1999;99:451–454. - PubMed
-
- Jones PA, Laird PW. Cancer epigenetics comes of age. Nature genetics. 1999;21:163–167. - PubMed
-
- Lund AH, van Lohuizen M. Epigenetics and cancer. Genes & development. 2004;18:2315–2335. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
