

Proteasome nuclear activity affects chromosome stability by controlling the turnover of Mms22, a protein important for DNA repair
- PMID: 20174551
- PMCID: PMC2824753
- DOI: 10.1371/journal.pgen.1000852
Proteasome nuclear activity affects chromosome stability by controlling the turnover of Mms22, a protein important for DNA repair
Abstract
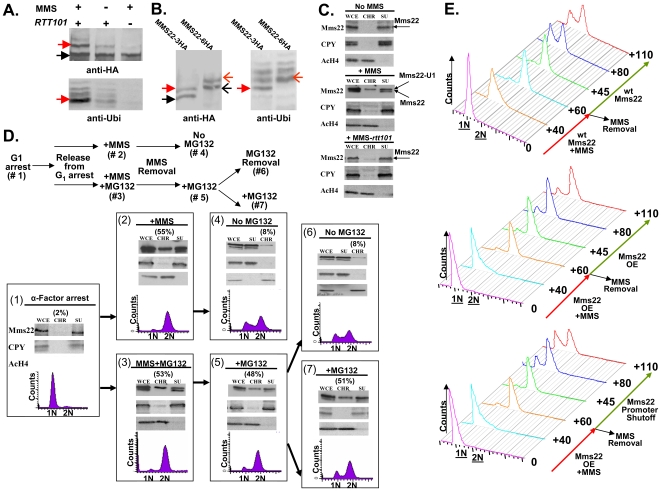
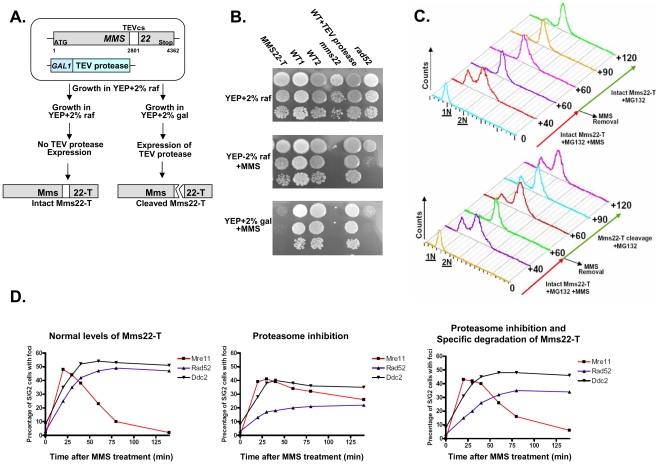
To expand the known spectrum of genes that maintain genome stability, we screened a recently released collection of temperature sensitive (Ts) yeast mutants for a chromosome instability (CIN) phenotype. Proteasome subunit genes represented a major functional group, and subsequent analysis demonstrated an evolutionarily conserved role in CIN. Analysis of individual proteasome core and lid subunit mutations showed that the CIN phenotype at semi-permissive temperature is associated with failure of subunit localization to the nucleus. The resultant proteasome dysfunction affects chromosome stability by impairing the kinetics of double strand break (DSB) repair. We show that the DNA repair protein Mms22 is required for DSB repair, and recruited to chromatin in a ubiquitin-dependent manner as a result of DNA damage. Moreover, subsequent proteasome-mediated degradation of Mms22 is necessary and sufficient for cell cycle progression through the G(2)/M arrest induced by DNA damage. Our results demonstrate for the first time that a double strand break repair protein is a proteasome target, and thus link nuclear proteasomal activity and DSB repair.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
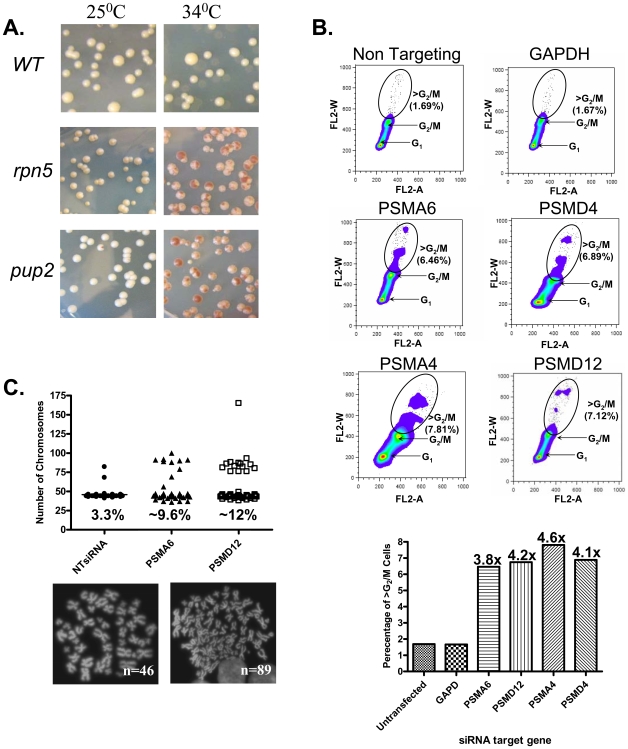
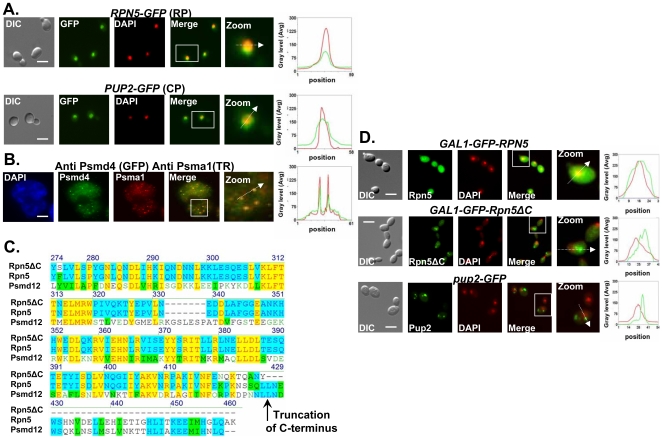
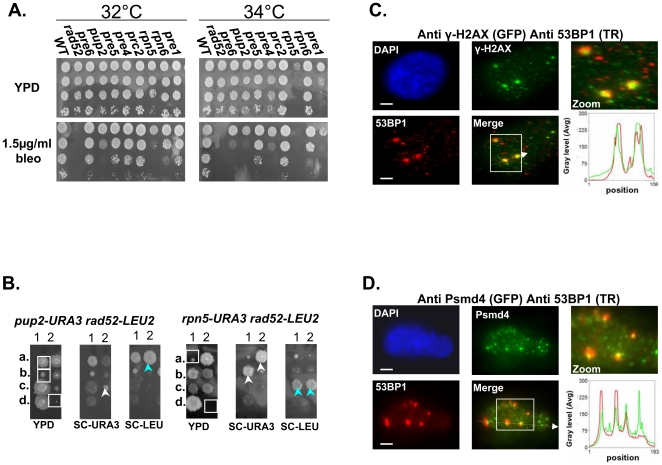
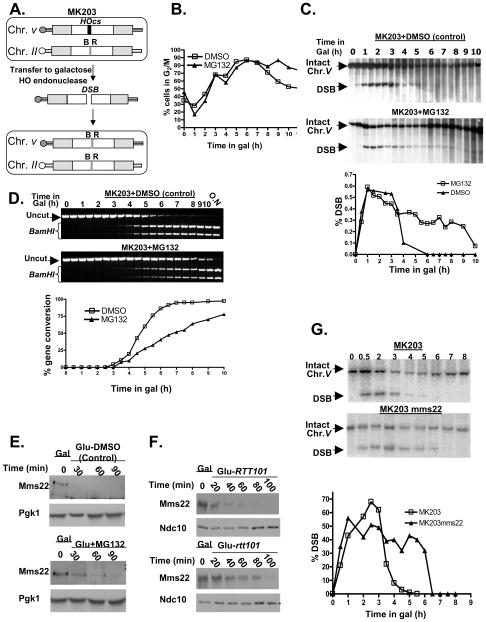
References
-
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. - PubMed
-
- Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst) 2007;6:994–1003. - PubMed
-
- Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
