

Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/-) mouse model
- PMID: 20174578
- PMCID: PMC2824822
- DOI: 10.1371/journal.pone.0009298
Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/-) mouse model
Abstract
Background: Loss-of-function mutations in SCN5A, the gene encoding Na(v)1.5 Na+ channel, are associated with inherited cardiac conduction defects and Brugada syndrome, which both exhibit variable phenotypic penetrance of conduction defects. We investigated the mechanisms of this heterogeneity in a mouse model with heterozygous targeted disruption of Scn5a (Scn5a(+/-) mice) and compared our results to those obtained in patients with loss-of-function mutations in SCN5A.
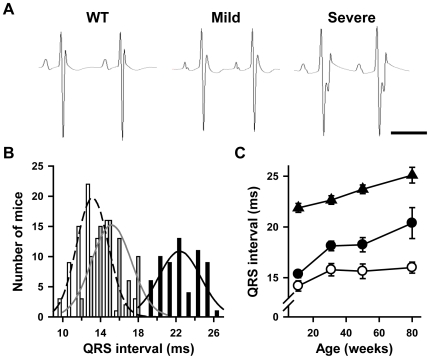
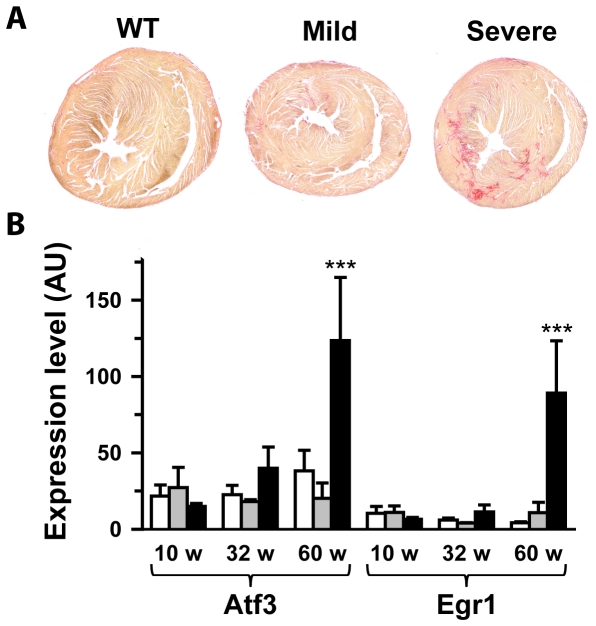
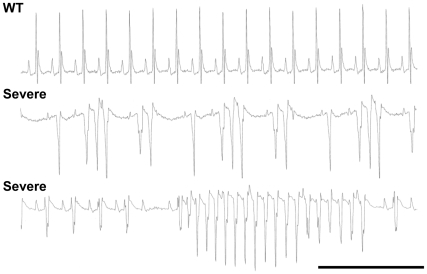
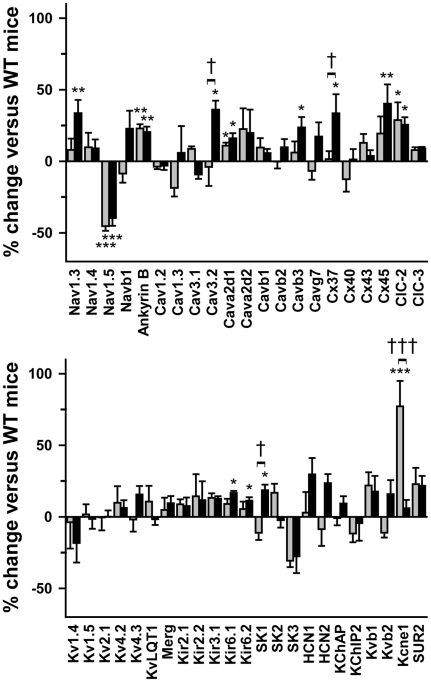
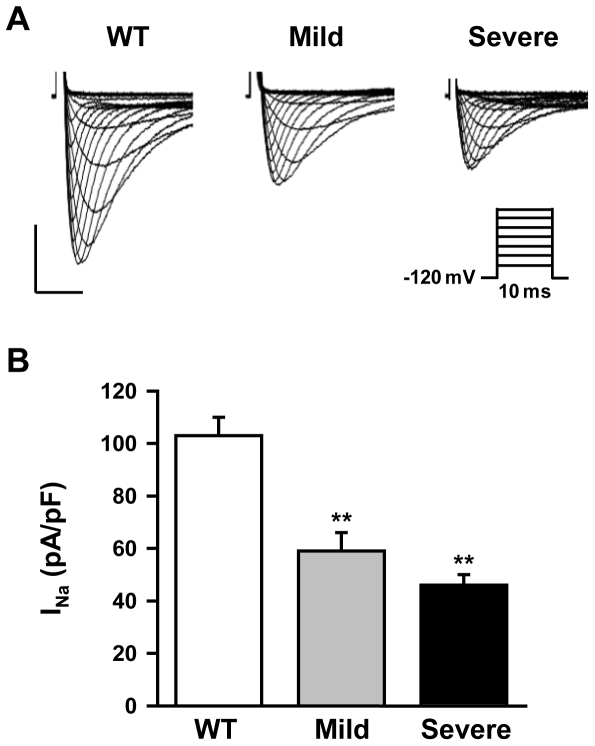
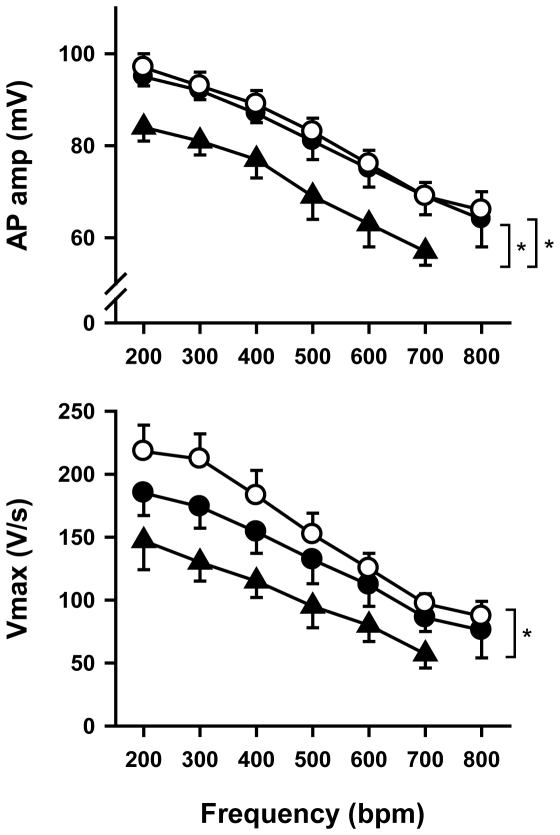
Methodology/principal findings: Based on ECG, 10-week-old Scn5a(+/-) mice were divided into 2 subgroups, one displaying severe ventricular conduction defects (QRS interval>18 ms) and one a mild phenotype (QRS< or = 18 ms; QRS in wild-type littermates: 10-18 ms). Phenotypic difference persisted with aging. At 10 weeks, the Na+ channel blocker ajmaline prolonged QRS interval similarly in both groups of Scn5a(+/-) mice. In contrast, in old mice (>53 weeks), ajmaline effect was larger in the severely affected subgroup. These data matched the clinical observations on patients with SCN5A loss-of-function mutations with either severe or mild conduction defects. Ventricular tachycardia developed in 5/10 old severely affected Scn5a(+/-) mice but not in mildly affected ones. Correspondingly, symptomatic SCN5A-mutated Brugada patients had more severe conduction defects than asymptomatic patients. Old severely affected Scn5a(+/-) mice but not mildly affected ones showed extensive cardiac fibrosis. Mildly affected Scn5a(+/-) mice had similar Na(v)1.5 mRNA but higher Na(v)1.5 protein expression, and moderately larger I(Na) current than severely affected Scn5a(+/-) mice. As a consequence, action potential upstroke velocity was more decreased in severely affected Scn5a(+/-) mice than in mildly affected ones.
Conclusions: Scn5a(+/-) mice show similar phenotypic heterogeneity as SCN5A-mutated patients. In Scn5a(+/-) mice, phenotype severity correlates with wild-type Na(v)1.5 protein expression.
Conflict of interest statement
Figures


References
-
- Schott J-J, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–21. - PubMed
-
- Probst V, Kyndt F, Potet F, Trochu J-N, Mialet G, et al. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenegre disease. J Am Coll Cardiol. 2003;41:643–652. - PubMed
-
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. - PubMed
-
- Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003;57:961–973. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
