

A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction
- PMID: 20175916
- PMCID: PMC2838872
- DOI: 10.1186/1471-2105-11-99
A new protein binding pocket similarity measure based on comparison of clouds of atoms in 3D: application to ligand prediction
Abstract
Background: Predicting which molecules can bind to a given binding site of a protein with known 3D structure is important to decipher the protein function, and useful in drug design. A classical assumption in structural biology is that proteins with similar 3D structures have related molecular functions, and therefore may bind similar ligands. However, proteins that do not display any overall sequence or structure similarity may also bind similar ligands if they contain similar binding sites. Quantitatively assessing the similarity between binding sites may therefore be useful to propose new ligands for a given pocket, based on those known for similar pockets.
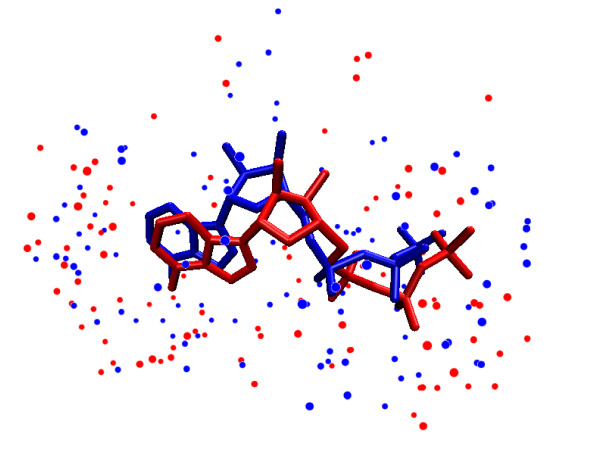
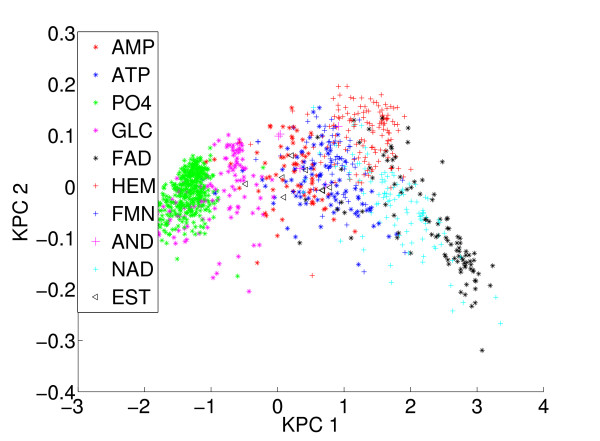
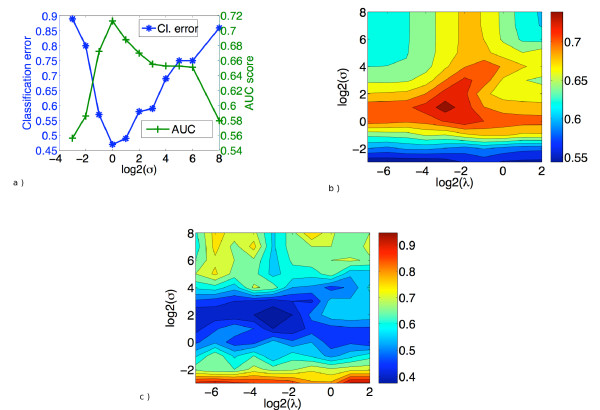
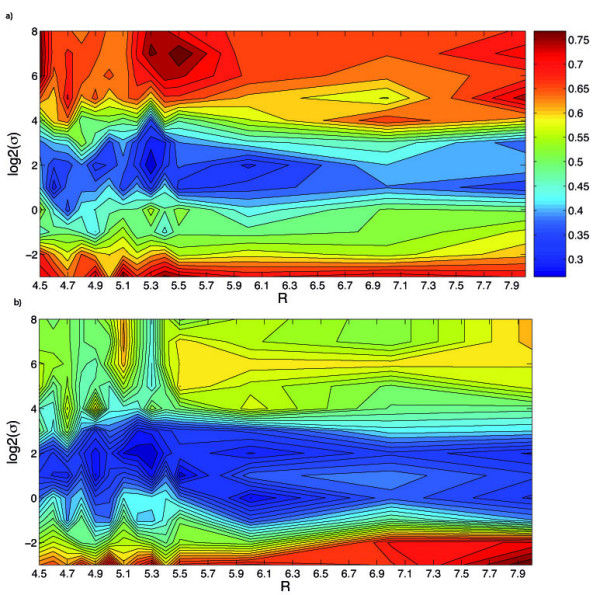
Results: We propose a new method to quantify the similarity between binding pockets, and explore its relevance for ligand prediction. We represent each pocket by a cloud of atoms, and assess the similarity between two pockets by aligning their atoms in the 3D space and comparing the resulting configurations with a convolution kernel. Pocket alignment and comparison is possible even when the corresponding proteins share no sequence or overall structure similarities. In order to predict ligands for a given target pocket, we compare it to an ensemble of pockets with known ligands to identify the most similar pockets. We discuss two criteria to evaluate the performance of a binding pocket similarity measure in the context of ligand prediction, namely, area under ROC curve (AUC scores) and classification based scores. We show that the latter is better suited to evaluate the methods with respect to ligand prediction, and demonstrate the relevance of our new binding site similarity compared to existing similarity measures.
Conclusions: This study demonstrates the relevance of the proposed method to identify ligands binding to known binding pockets. We also provide a new benchmark for future work in this field. The new method and the benchmark are available at http://cbio.ensmp.fr/paris/.
Figures



Similar articles
-
Estimation of important binding sites in compounds that interact with proteins.Comput Biol Chem. 2021 Aug;93:107511. doi: 10.1016/j.compbiolchem.2021.107511. Epub 2021 May 19. Comput Biol Chem. 2021. PMID: 34107451
-
PatchSurfers: Two methods for local molecular property-based binding ligand prediction.Methods. 2016 Jan 15;93:41-50. doi: 10.1016/j.ymeth.2015.09.026. Epub 2015 Sep 30. Methods. 2016. PMID: 26427548 Free PMC article.
-
Constructing patch-based ligand-binding pocket database for predicting function of proteins.BMC Bioinformatics. 2012 Mar 13;13 Suppl 2(Suppl 2):S7. doi: 10.1186/1471-2105-13-S2-S7. BMC Bioinformatics. 2012. PMID: 22536870 Free PMC article.
-
Compound activity prediction using models of binding pockets or ligand properties in 3D.Curr Top Med Chem. 2012;12(17):1869-82. doi: 10.2174/156802612804547335. Curr Top Med Chem. 2012. PMID: 23116466 Free PMC article. Review.
-
Chemogenomics in drug discovery: computational methods based on the comparison of binding sites.Future Med Chem. 2012 Oct;4(15):1971-9. doi: 10.4155/fmc.12.147. Future Med Chem. 2012. PMID: 23088277 Review.
Cited by
-
Binding ligand prediction for proteins using partial matching of local surface patches.Int J Mol Sci. 2010;11(12):5009-26. doi: 10.3390/ijms11125009. Epub 2010 Dec 6. Int J Mol Sci. 2010. PMID: 21614188 Free PMC article.
-
PL-PatchSurfer: a novel molecular local surface-based method for exploring protein-ligand interactions.Int J Mol Sci. 2014 Aug 27;15(9):15122-45. doi: 10.3390/ijms150915122. Int J Mol Sci. 2014. PMID: 25167137 Free PMC article.
-
Simple Ligand-Receptor Interaction Descriptor (SILIRID) for alignment-free binding site comparison.Comput Struct Biotechnol J. 2014 Jun 11;10(16):33-7. doi: 10.1016/j.csbj.2014.05.004. eCollection 2014 Jun. Comput Struct Biotechnol J. 2014. PMID: 25210596 Free PMC article.
-
Surface-based protein binding pocket similarity.Proteins. 2011 Sep;79(9):2746-63. doi: 10.1002/prot.23103. Epub 2011 Jul 18. Proteins. 2011. PMID: 21769944 Free PMC article.
-
DARC: Mapping Surface Topography by Ray-Casting for Effective Virtual Screening at Protein Interaction Sites.J Med Chem. 2016 May 12;59(9):4152-70. doi: 10.1021/acs.jmedchem.5b00150. Epub 2015 Jul 10. J Med Chem. 2016. PMID: 26126123 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
