

Poly(ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by alpha-tocopherol and MEK inhibition
- PMID: 20177052
- PMCID: PMC2857103
- DOI: 10.1074/jbc.M109.044206
Poly(ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by alpha-tocopherol and MEK inhibition
Abstract
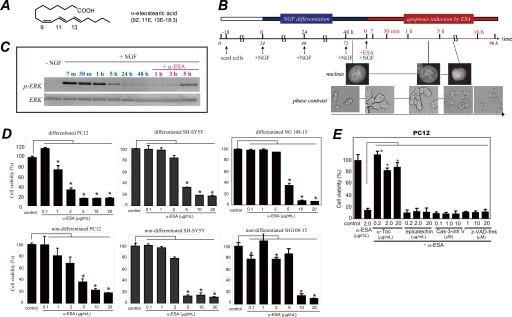
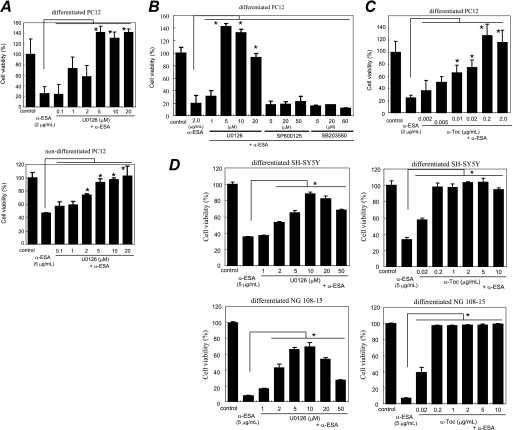
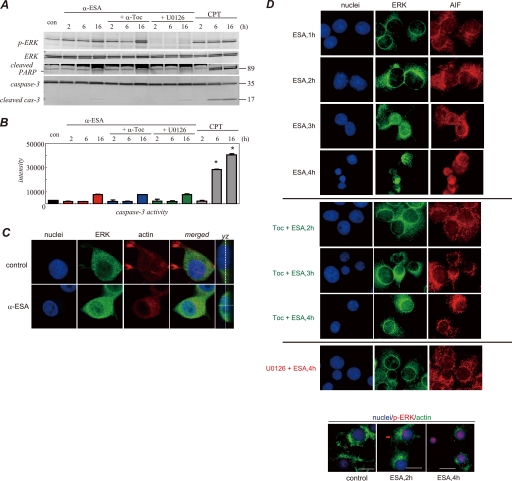
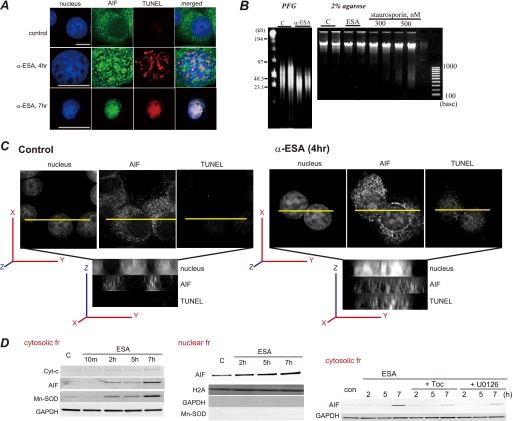
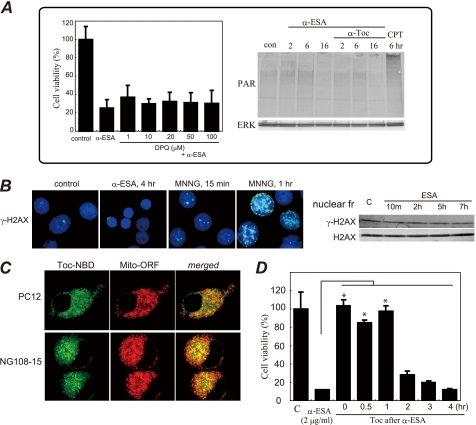
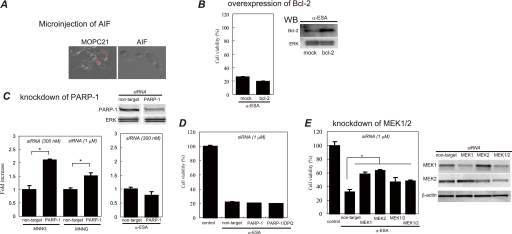
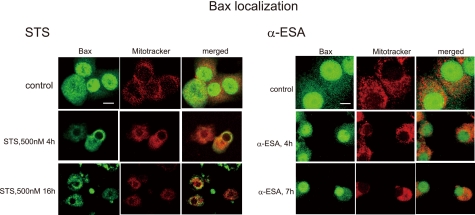
Poly(ADP-ribose)polymerase-1 (PARP-1) is thought to be required for apoptosis-inducing factor (AIF) release from mitochondria in caspase-independent apoptosis. The mechanism by which AIF is released through PARP-1 remains unclear. Here, we provide evidence that PARP-1-independent AIF release and cell death are induced by a trienoic fatty acid, alpha-eleostearic acid (alpha-ESA). Alpha-ESA induced the caspase-independent and AIF-initiated apoptotic death of neuronal cell lines, independently of PARP-1 activation. The cell death was inhibited by the MEK inhibitor U0126 and by knockdown of MEK using small interfering RNA. However, inhibitors for JNK, p38 inhibitors, calpain, phospholipase A(2), and phosphatidylinositol 3-kinase, did not block cell death. AIF was translocated to the nucleus after the induction of apoptosis by alpha-ESA in differentiated PC12 cells without activating caspase-3 and PARP-1. The alpha-ESA-mediated cell death was not inhibited by PARP inhibitor 3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinoline and by knockdown of PARP-1 using small interfering RNA. Unlike N-methyl-N'-nitro-N-nitrosoguanidine treatment, histone-phosphorylated histone 2AX was not phosphorylated by alpha-ESA, which suggests no DNA damage. Overexpression of Bcl-2 did not inhibit the cell death. alpha-ESA caused a small quantity of superoxide production in the mitochondria, resulting in the reduction of mitochondrial membrane potential, both of which were blocked by a trace amount of alpha-tocopherol localized in the mitochondria. Our results demonstrate that alpha-ESA induces PARP-1-independent AIF release and cell death without activating Bax, cytochrome c, and caspase-3. MEK is also a key molecule, although the link between ERK, AIF release, and cell death remains unknown. Finding molecules that regulate AIF release may be an important therapeutic target for the treatment of neuronal injury.
Figures


Similar articles
-
Eleostearic acid induces RIP1-mediated atypical apoptosis in a kinase-independent manner via ERK phosphorylation, ROS generation and mitochondrial dysfunction.Cell Death Dis. 2013 Jun 20;4(6):e674. doi: 10.1038/cddis.2013.188. Cell Death Dis. 2013. PMID: 23788031 Free PMC article.
-
Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis.Mol Cell Biol. 2007 Jul;27(13):4844-62. doi: 10.1128/MCB.02141-06. Epub 2007 Apr 30. Mol Cell Biol. 2007. PMID: 17470554 Free PMC article.
-
Diabetic cystopathy is associated with PARP/JNK/mitochondrial apoptotic pathway-mediated bladder apoptosis.Neurourol Urodyn. 2010 Sep;29(7):1332-7. doi: 10.1002/nau.20869. Neurourol Urodyn. 2010. PMID: 20879002
-
Deadly conversations: nuclear-mitochondrial cross-talk.J Bioenerg Biomembr. 2004 Aug;36(4):287-94. doi: 10.1023/B:JOBB.0000041755.22613.8d. J Bioenerg Biomembr. 2004. PMID: 15377859 Review.
-
Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos.Exp Neurol. 2009 Aug;218(2):193-202. doi: 10.1016/j.expneurol.2009.03.020. Epub 2009 Mar 28. Exp Neurol. 2009. PMID: 19332058 Free PMC article. Review.
Cited by
-
The Sound of Silence: RNAi in Poly (ADP-Ribose) Research.Genes (Basel). 2012 Dec 6;3(4):779-805. doi: 10.3390/genes3040779. Genes (Basel). 2012. PMID: 24705085 Free PMC article.
-
Effects of folic acid on the antiproliferative efficiency of doxorubicin, camptothecin and methyl methanesulfonate in MCF-7 cells by mRNA endpoints.Saudi J Biol Sci. 2018 Dec;25(8):1568-1576. doi: 10.1016/j.sjbs.2016.02.005. Epub 2016 Feb 10. Saudi J Biol Sci. 2018. PMID: 30581319 Free PMC article.
-
NSP 5a3a: a potential novel cancer target in head and neck carcinoma.Oncotarget. 2010 Oct;1(6):423-435. doi: 10.18632/oncotarget.176. Oncotarget. 2010. PMID: 21311098 Free PMC article.
-
α-Tocopherol at Nanomolar Concentration Protects Cortical Neurons against Oxidative Stress.Int J Mol Sci. 2017 Jan 21;18(1):216. doi: 10.3390/ijms18010216. Int J Mol Sci. 2017. PMID: 28117722 Free PMC article.
-
α-Tocopherol at nanomolar concentration protects PC12 cells from hydrogen peroxide-induced death and modulates protein kinase activities.Int J Mol Sci. 2012;13(9):11543-11568. doi: 10.3390/ijms130911543. Epub 2012 Sep 14. Int J Mol Sci. 2012. PMID: 23109870 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
